Synthetic Cannabinoids Induce Acute Lung Inflammation Via Cannabinoid Receptor 1 Activation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Marijuana: Drug of Abuse Or Therapeutic Option? Learning Objectives
Handout for the Neuroscience Education Institute (NEI) online activity: Marijuana: Drug of Abuse or Therapeutic Option? Learning Objectives • Explain how cannabinoids affect the body and the brain • Educate patients about: – Evidence of efficacy for mental health and other conditions – Potential risks of cannabis use Timeline 1989? 90?: Discovery of ~2000 BC: binding site 2015: Elimination of Chinese 1943: Marijuana for THC— 1992: US Public Health emperors removed from CB1 Endogenous Service oversight cannabinoid recommend listing as a 1963: 1964: THC receptor for obtaining anandamide marijuana as medication in US Cannabidiol isolated marijuana for discovered medicine Pharmacopeia isolated research purposes 1995: 1851: Marijuana 1961: United 1970: Marijuana is Endogenous Aug 11 2016: listed as a Nations Single labeled Schedule I cannabinoid 2-AG DEA declines to medication in US Convention on by the US discovered reschedule Pharmacopeia Narcotic Drugs: Substance Abuse marijuana marijuana said to Act; this restricts be dangerous with both personal use no medical value and access for research purposes THE INTERSECTION OF THE HEALTHCARE AND CANNABIS INDUSTRIES What is Cannabis? 500 chemicals 100 cannabinoids Best understood: THC and CBD Scheduling of Controlled Substances No medicinal value, high Moderate to Lower High potential Low potential potential for low potential potential for for abuse for abuse abuse for abuse abuse Schedule I Schedule II Schedule III Schedule IV Schedule V Marijuana Cocaine Tylenol w/ Tramadol Robitussin AC Heroin -

Cannabinoid Receptor and Inflammation
Cannabinoid Receptor and Inflammation Newman Osafo1, Oduro Yeboah1, Aaron Antwi1, and George Ainooson1 1Kwame Nkrumah University of Science and Technology September 11, 2020 Abstract The eventual discovery of endogenous cannabinoid receptors CB1 and CB2 and their endogenous ligands has generated interest with regards to finally understanding the endocannabinoid system. Its role in the normal physiology of the body and its implication in pathological states such as cardiovascular diseases, neoplasm, depression and pain have been subjects of scientific interest. In this review the authors focus on the endogenous cannabinoid pathway, the critical role of cannabinoid receptors in signaling and mediation of neurodegeneration and other inflammatory responses as well as its potential as a drug target in the amelioration of some inflammatory conditions. Though the exact role of the endocannabinoid system is not fully understood, the evidence found leans heavily towards a great potential in exploiting both its central and peripheral pathways in disease management. Cannabinoid therapy has already shown great promise in several preclinical and clinical trials. 1.0 Introduction Ethnopharmacological studies have shown the use of Cannabis sativa in traditional medicine for over a thousand years, with its widespread use promoted by its psychotropic effects (McCoy, 2016; Turcotte et al., 2016). The discovery of a receptor within human body, that is selectively activated by cannabinoids suggested the presence of at least one endogenous ligand for this receptor. This is confirmed by the discovery of two endogenously synthesized lipid mediators, 2-arachidonoyl-glycerol and arachidonoylethanolamide, which function as high-affinity ligands for a subfamily of cannabinoid receptors ubiquitously distributed in the central nervous system, known as the CB1 receptors (Turcotte et al., 2016). -

Cannabinoid Interventions for PTSD: Where to Next?
Accepted Manuscript Cannabinoid interventions for PTSD: Where to next? Luke Ney, Allison Matthews, Raimondo Bruno, Kim Felmingham PII: S0278-5846(19)30034-X DOI: https://doi.org/10.1016/j.pnpbp.2019.03.017 Reference: PNP 9622 To appear in: Progress in Neuropsychopharmacology & Biological Psychiatry Received date: 14 January 2019 Revised date: 20 March 2019 Accepted date: 29 March 2019 Please cite this article as: L. Ney, A. Matthews, R. Bruno, et al., Cannabinoid interventions for PTSD: Where to next?, Progress in Neuropsychopharmacology & Biological Psychiatry, https://doi.org/10.1016/j.pnpbp.2019.03.017 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT 1 Cannabinoid interventions for PTSD: Where to next? Luke Ney1, Allison Matthews1, Raimondo Bruno1 and Kim Felmingham2 1School of Psychology, University of Tasmania, Australia 2School of Psychological Sciences, University of Melbourne, Australia ACCEPTED MANUSCRIPT ACCEPTED MANUSCRIPT 2 Abstract Cannabinoids are a promising method for pharmacological treatment of post- traumatic stress disorder (PTSD). Despite considerable research devoted to the effect of cannabinoid modulation on PTSD symptomology, there is not a currently agreed way by which the cannabinoid system should be targeted in humans. In this review, we present an overview of recent research identifying neurological pathways by which different cannabinoid-based treatments may exert their effects on PTSD symptomology. -

Effects of Cannabinoids on the Development of Chick Embryos In
www.nature.com/scientificreports OPEN Efects of cannabinoids on the development of chick embryos in ovo Received: 16 April 2019 Sofa B. Gustafsson & Stig O. P. Jacobsson Accepted: 3 September 2019 We have examined the efects of the synthetic cannabinoids HU 210 and HU 211, the plant-derived Published online: 17 September 2019 cannabidiol and the endogenous cannabinoid anandamide on the viability and development of chick embryos. Fertilized White Leghorn chicken eggs were injected with the test compounds or carrier vehicle, via a drilled small hole in the egg, directly into the egg yolk. After nine days of exposure, the embryonal viability, length and wet weight of embryos, and wet weight of brains were measured, and the development stages were assessed according to the Hamburger and Hamilton (HH) scale. The potent synthetic cannabinoid receptor agonist HU 210 and the non-psychotropic cannabidiol were embryotoxic at the highest concentrations examined (10 µM and 50 µM, respectively), with no viable embryos after the HU 210 injection, and 20% viability after the cannabidiol injections. The efects of HU 210 on the chick embryo were attenuated by α-tocopherol and the cannabinoid receptor antagonist AM251, whereas only α-tocopherol gave a statistically signifcant protection against the embryotoxic efects of cannabidiol. This study shows that exposure to plant-derived or synthetic cannabinoids during early embryonal development decreases embryonal viability. Extrapolation of data across species is of course difcult, but the data would argue against the use of cannabinoids, be it recreationally or therapeutically, during pregnancy. Cannabis has a long history of medical and therapeutic use, and the discovery of the cannabinoid (CB) signalling system, comprising CB receptors, endogenous ligands and enzymes for ligand biosynthesis and inactivation, has led to an enormous expansion of the CB research feld and in turn to the identifcation of new targets for therapeutic drugs1–3. -

The Use of Cannabinoids in Animals and Therapeutic Implications for Veterinary Medicine: a Review
Veterinarni Medicina, 61, 2016 (3): 111–122 Review Article doi: 10.17221/8762-VETMED The use of cannabinoids in animals and therapeutic implications for veterinary medicine: a review L. Landa1, A. Sulcova2, P. Gbelec3 1Faculty of Medicine, Masaryk University, Brno, Czech Republic 2Central European Institute of Technology, Masaryk University, Brno, Czech Republic 3Veterinary Hospital and Ambulance AA Vet, Prague, Czech Republic ABSTRACT: Cannabinoids/medical marijuana and their possible therapeutic use have received increased atten- tion in human medicine during the last years. This increased attention is also an issue for veterinarians because particularly companion animal owners now show an increased interest in the use of these compounds in veteri- nary medicine. This review sets out to comprehensively summarise well known facts concerning properties of cannabinoids, their mechanisms of action, role of cannabinoid receptors and their classification. It outlines the main pharmacological effects of cannabinoids in laboratory rodents and it also discusses examples of possible beneficial use in other animal species (ferrets, cats, dogs, monkeys) that have been reported in the scientific lit- erature. Finally, the article deals with the prospective use of cannabinoids in veterinary medicine. We have not intended to review the topic of cannabinoids in an exhaustive manner; rather, our aim was to provide both the scientific community and clinical veterinarians with a brief, concise and understandable overview of the use of cannabinoids in veterinary -

Pharmacodynamics of Cannabinoids
Open Access Archives of Pharmacy and Pharmaceutical Sciences Review Article Pharmacodynamics of cannabinoids Alexandra Sulcova* ISSN ICCI - International Cannabis and Cannabinoids Institute, Jachymova 26/2, 110 00 Praha, 2639-992X Czech Republic “Pharmacodynamics of cannabinoids “(i.e. a set of biological effects elicited in the *Address for Correspondence: Alexandra Sulcova, M.D, Ph.D, Professor of Pharmacology, living organism by interaction with its biochemical and biophysical functions up to the FCMA, FECNP, FCINP, ICCI - International cellular level) is studied for a long time during both, physiological and pathological Cannabis and Cannabinoids Institute, Jachymova conditions. Cannabinoids received their names according to their natural occurrence 26/2, 110 00 Praha, Czech Republic, Tel: 420 732167678; Email: [email protected] as constituents of Cannabis sativa L. (marijuana). The species was classiied in the “Linnaeus’s Species Plantarum (1753)”, the word “sativa” means things that are Submitted: 12 April 2019 Approved: 07 May 2019 cultivated [1]. For ages, people have used cannabis-based preparations for healing and Published: 08 May 2019 pain suppression until the discovery (in 1897) of aspirin (acetylsalicylic acid) which contemporary medicine uses until today. Chemical investigation of marijuana conirmed Copyright: © 2019 Sulcova A. This is an open access article distributed under the Creative various cannabinoid-type components called cannabinoids (presently estimated at Commons Attribution License, which permits about 150). Regarding their possible pharmacodynamic effects, tetrahydrocannabinol unrestricted use, distribution, and reproduction (THC) and cannabidiol (CBD) are the most explored. The determination of THC structure in any medium, provided the original work is properly cited by means of nuclear magnetic resonance imaging increased sharply the number of professional scientiic reports dealing with the studies of THC pharmacodynamic mechanisms of action [2]. -

Cannabinoid Treatment of Opiate Addiction
Neuroanatomy and Behaviour, 2021, 3(1), e14. Neuroanatomy ISSN: 2652-1768 and H doi: 10.35430/nab.2021.e14 ε Behaviour REVIEW Cannabinoid treatment of opiate addiction Erin McLemon1 and Rose Chesworth1,* 1School of Medicine, Western Sydney University, Campbelltown NSW 2560, Australia *[email protected] Abstract Opioid abuse is a growing global problem. Current therapies for opioid abuse target withdrawal symptoms and have several adverse side effects. There are no treatments to address opioid-induced neural adaptations associated with abuse and addiction. Preclinical research demonstrates interactions between the endogenous opioid and cannabinoid systems, suggesting that cannabinoids may be used to treat opioid addiction and dependence. The aim of this review is to assess how cannabinoids affect behavioural and molecular measures of opioid dependence and addiction-like behaviour in animal models. It appears that cannabidiol and cannabinoid receptor 1 (CB1R) antagonists have potential for treating drug-craving and drug-seeking behaviour, based on evidence from preclinical animal models. Ligands which inhibit the action of cannabinoid degradation enzymes also show promise in reducing opioid withdrawal symptoms and opioid self-administration in rodents. Agonists of CB1R could be useful for treating symptoms of opioid withdrawal; however, the clinical utility of these drugs is limited by side effects, the potential for cannabinoid addiction and an increase in opiate tolerance induced by cannabinoid consumption. The mechanisms by which cannabinoids reduce opioid addiction-relevant behaviours include modulation of cannabinoid, serotonin, and dopamine receptors, as well as signalling cascades involving ERK-CREB-BDNF and peroxisome proliferator-activated receptor-a. Identifying the receptors involved and their mechanism of action remains a critical area of future research. -
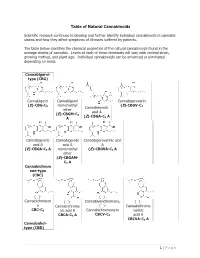
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic -

Cardiovascular Effects of Marijuana and Synthetic Cannabinoids: the Good, the Bad, and the Ugly
REVIEWS PREVENTION OF CVD Cardiovascular effects of marijuana and synthetic cannabinoids: the good, the bad, and the ugly Pal Pacher1, Sabine Steffens2, György Haskó3, Thomas H. Schindler4 and George Kunos5 Abstract | Dysregulation of the endogenous lipid mediators endocannabinoids and their G‑protein‑coupled cannabinoid receptors 1 and 2 (CB1R and CB2R) has been implicated in a variety of cardiovascular pathologies. Activation of CB1R facilitates the development of cardiometabolic disease, whereas activation of CB2R (expressed primarily in immune cells) exerts anti-inflammatory effects. The psychoactive constituent of marijuana, Δ9-tetrahydrocannabinol (THC), is an agonist of both CB1R and CB2R, and exerts its psychoactive and adverse cardiovascular effects through the activation of CB1R in the central nervous and cardiovascular systems. The past decade has seen a nearly tenfold increase in the THC content of marijuana as well as the increased availability of highly potent synthetic cannabinoids for recreational use. These changes have been accompanied by the emergence of serious adverse cardiovascular events, including myocardial infarction, cardiomyopathy, arrhythmias, stroke, and cardiac arrest. In this Review, we summarize the role of the endocannabinoid system in cardiovascular disease, and critically discuss the cardiovascular consequences of marijuana and synthetic cannabinoid use. With the legalization of marijuana for medicinal purposes and/or recreational use in many countries, physicians should be alert to the possibility -

WO 2018/152334 Al 23 August 2018 (23.08.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/152334 Al 23 August 2018 (23.08.2018) W !P O PCT (51) International Patent Classification: (US). YUCEL, Tuna; 28 Monmouth Avenue, Medford, A61K 9/107 (2006.01) A61K 31/352 (2006.01) MA 02155 (US). BOYLAN, Nicholas, J.; 215 Green A61K 47/26 (2006.01) A61K 9/48 (2006.01) Street, Boylston, MA 01505 (US). A61K 47/14 (2006.01) A61K 9/00 (2006.01) (74) Agent: EISENSCHENK, Frank, C. et al; Saliwanchik, A61K 31/05 (2006 .01) A61P 25/06 (2006 .0 1) Lloyd & Eisenschenk, P.O. Box 142950, Gainesville, FL (21) International Application Number: 32614-2950 (US). PCT/US2018/018382 (81) Designated States (unless otherwise indicated, for every (22) International Filing Date: kind of national protection available): AE, AG, AL, AM, 15 February 2018 (15.02.2018) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (25) Filing Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (26) Publication Language: English HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (30) Priority Data: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 62/459,086 15 February 2017 (15.02.2017) OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, 62/546,149 16 August 2017 (16.08.2017) SC, SD, SE, SG, SK, SL, SM, ST, SV, SY,TH, TJ, TM, TN, (71) Applicant: MOLECULAR INFUSIONS, LLC [US/US]; TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Are Cannabidiol and Δ9‐Tetrahydrocannabivarin Negative
British Journal of DOI:10.1111/bph.12944 www.brjpharmacol.org BJP Pharmacology REVIEW Correspondence Dr John M McPartland, 53 Washington Street Ext., Middlebury, VT 05753, USA. Are cannabidiol and E-mail: [email protected] 9 ---------------------------------------------------------------- Δ Received -tetrahydrocannabivarin 21 February 2014 Revised 12 September 2014 negative modulators of the Accepted endocannabinoid system? A 16 September 2014 systematic review John M McPartland1, Marnie Duncan2, Vincenzo Di Marzo3 and Roger G Pertwee4 1Division of Molecular Biology, GW Pharmaceuticals, Salisbury, Wiltshire, UK, 2GW Pharmaceuticals, Porton Down Science Park, Salisbury, Wiltshire, UK, 3Endocannabinoid Research Group, Institute of Biomolecular Chemistry, CNR, Napoli, Italy, and 4School of Medical Sciences, Institute of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen, UK Based upon evidence that the therapeutic properties of Cannabis preparations are not solely dependent upon the presence of Δ9-tetrahydrocannabinol (THC), pharmacological studies have been recently carried out with other plant cannabinoids (phytocannabinoids), particularly cannabidiol (CBD) and Δ9-tetrahydrocannabivarin (THCV). Results from some of these studies have fostered the view that CBD and THCV modulate the effects of THC via direct blockade of cannabinoid CB1 receptors, thus behaving like first-generation CB1 receptor inverse agonists, such as rimonabant. Here, we review in vitro and ex vivo mechanistic studies of CBD and THCV, and synthesize -

CB2 Cannabinoid Receptor Agonist Enantiomers HU-433 and HU-308: an Inverse Relationship Between Binding Affinity and Biological Potency
CB2 cannabinoid receptor agonist enantiomers HU-433 and HU-308: An inverse relationship between binding affinity and biological potency Reem Smouma, Saja Baraghithyb, Mukesh Chourasiaa,c,1, Aviva Breuera, Naama Mussaib, Malka Attar-Namdarb, Natalya M. Koganb, Bitya Raphaelb, Daniele Bologninid,e, Maria G. Casciod, Pietro Marinid, Roger G. Pertweed, Avital Shurkia,c, Raphael Mechoulama,2, and Itai Babb,3 aInstitute for Drug Research, Hebrew University of Jerusalem, Jerusalem 91120, Israel; bBone Laboratory, Hebrew University of Jerusalem, Jerusalem 91120, Israel; cThe Lise-Meitner Minerva Center for Computational Quantum Chemistry, Hebrew University of Jerusalem, Jerusalem 91120, Israel; dInstitute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD, Scotland, United Kingdom; and eMolecular Pharmacology Group, Institute of Molecular, Cell and System Biology, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow G12 8QQ, Scotland, United Kingdom Edited by Leslie Lars Iversen, University of Oxford, Oxford, United Kingdom, and approved June 9, 2015 (received for review February 23, 2015) Activation of the CB2 receptor is apparently an endogenous protec- by using concentrations in the nanomolar range (2, 14). Surprisingly, tive mechanism. Thus, it restrains inflammation and protects the testing a new HU-308 batch, we noticed a 3- to 4-magnitude de- skeleton against age-related bone loss. However, the endogenous crease in the dose that triggers optimal proliferative response in cannabinoids, as well as Δ9-tetrahydrocannabinol, the main plant osteoblasts, reasoning that this preparation contained a significant psychoactive constituent, activate both cannabinoid receptors, CB1 amount of the HU-308 enantiomer, presumably responsible for the and CB2. HU-308 was among the first synthetic, selective CB2 ago- enhanced activity.