Nngv3n5-Issue-Text-Proof.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -
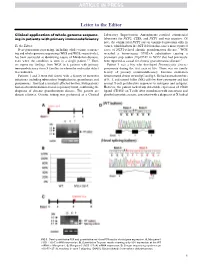
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Genome-Wide Association Identifies Candidate Genes That Influence The
Genome-wide association identifies candidate genes that influence the human electroencephalogram Colin A. Hodgkinsona,1, Mary-Anne Enocha, Vibhuti Srivastavaa, Justine S. Cummins-Omana, Cherisse Ferriera, Polina Iarikovaa, Sriram Sankararamanb, Goli Yaminia, Qiaoping Yuana, Zhifeng Zhoua, Bernard Albaughc, Kenneth V. Whitea, Pei-Hong Shena, and David Goldmana aLaboratory of Neurogenetics, National Institute on Alcohol Abuse and Alcoholism, Rockville, MD 20852; bComputer Science Department, University of California, Berkeley, CA 94720; and cCenter for Human Behavior Studies, Weatherford, OK 73096 Edited* by Raymond L. White, University of California, Emeryville, CA, and approved March 31, 2010 (received for review July 23, 2009) Complex psychiatric disorders are resistant to whole-genome reflects rhythmic electrical activity of the brain. EEG patterns analysis due to genetic and etiological heterogeneity. Variation in dynamically and quantitatively index cortical activation, cognitive resting electroencephalogram (EEG) is associated with common, function, and state of consciousness. EEG traits were among the complex psychiatric diseases including alcoholism, schizophrenia, original intermediate phenotypes in neuropsychiatry, having been and anxiety disorders, although not diagnostic for any of them. EEG first recorded in humans in 1924 by Hans Berger, who documented traits for an individual are stable, variable between individuals, and the α rhythm, seen maximally during states of relaxation with eyes moderately to highly heritable. Such intermediate phenotypes closed, and supplanted by faster β waves during mental activity. appear to be closer to underlying molecular processes than are EEG can be used clinically for the evaluation and differential di- clinical symptoms, and represent an alternative approach for the agnosis of epilepsy and sleep disorders, differentiation of en- identification of genetic variation that underlies complex psychiat- cephalopathy from catatonia, assessment of depth of anesthesia, ric disorders. -

A Pathogenic Ctbp1 Missense Mutation Causes Altered Cofactor Binding and Transcriptional Activity
neurogenetics (2019) 20:129–143 https://doi.org/10.1007/s10048-019-00578-1 ORIGINAL ARTICLE A pathogenic CtBP1 missense mutation causes altered cofactor binding and transcriptional activity David B. Beck1 & T. Subramanian2 & S. Vijayalingam2 & Uthayashankar R. Ezekiel3 & Sandra Donkervoort4 & Michele L. Yang5 & Holly A. Dubbs6 & Xilma R. Ortiz-Gonzalez7 & Shenela Lakhani8 & Devorah Segal9 & Margaret Au10 & John M. Graham Jr10 & Sumit Verma11 & Darrel Waggoner12 & Marwan Shinawi13 & Carsten G. Bönnemann4 & Wendy K. Chung14 & G. Chinnadurai2 Received: 23 October 2018 /Revised: 18 March 2019 /Accepted: 9 April 2019 /Published online: 30 April 2019 # Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract We previously reported a pathogenic de novo p.R342W mutation in the transcriptional corepressor CTBP1 in four independent patients with neurodevelopmental disabilities [1]. Here, we report the clinical phenotypes of seven additional individuals with the same recurrent de novo CTBP1 mutation. Within this cohort, we identified consistent CtBP1-related phenotypes of intellectual disability, ataxia, hypotonia, and tooth enamel defects present in most patients. The R342W mutation in CtBP1 is located within a region implicated in a high affinity-binding cleft for CtBP-interacting proteins. Unbiased proteomic analysis demonstrated reduced interaction of several chromatin-modifying factors with the CtBP1 W342 mutant. Genome-wide transcriptome analysis in human glioblastoma cell lines expressing -CtBP1 R342 (wt) or W342 mutation revealed changes in the expression profiles of genes controlling multiple cellular processes. Patient-derived dermal fibroblasts were found to be more sensitive to apoptosis during acute glucose deprivation compared to controls. Glucose deprivation strongly activated the BH3-only pro-apoptotic gene NOXA, suggesting a link between enhanced cell death and NOXA expression in patient fibroblasts. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Assessment of Hematopoietic Failure Due to Rpl11 Deficiency in a Zebrafish Model of Diamond-Blackfan Anemia by Deep Sequencing
Zhang et al. BMC Genomics 2013, 14:896 http://www.biomedcentral.com/1471-2164/14/896 RESEARCH ARTICLE Open Access Assessment of hematopoietic failure due to Rpl11 deficiency in a zebrafish model of Diamond- Blackfan anemia by deep sequencing Zhaojun Zhang1†, Haibo Jia2†, Qian Zhang1, Yang Wan3, Yang Zhou2, Qiong Jia2, Wanguang Zhang4, Weiping Yuan3, Tao Cheng3, Xiaofan Zhu3* and Xiangdong Fang1* Abstract Background: Diamond–Blackfan anemia is a rare congenital red blood cell dysplasia that develops soon after birth. RPL11 mutations account for approximately 4.8% of human DBA cases with defective hematopoietic phenotypes. However, the mechanisms by which RPL11 regulates hematopoiesis in DBA remain elusive. In this study, we analyzed the transcriptome using deep sequencing data from an Rpl11-deficient zebrafish model to identify Rpl11-mediated hematopoietic failure and investigate the underlying mechanisms. Results: We characterized hematological defects in Rpl11-deficient zebrafish embryos by identifying affected hematological genes, hematopoiesis-associated pathways, and regulatory networks. We found that hemoglobin biosynthetic and hematological defects in Rpl11-deficient zebrafish were related to dysregulation of iron metabolism-related genes, including tfa, tfr1b, alas2 and slc25a37, which are involved in heme and hemoglobin biosynthesis. In addition, we found reduced expression of the hematopoietic stem cells (HSC) marker cmyb and HSC transcription factors tal1 and hoxb4a in Rpl11-deficient zebrafish embryos, indicating that the hematopoietic defects may be related to impaired HSC formation, differentiation, and proliferation. However, Rpl11 deficiency did not affect the development of other blood cell lineages such as granulocytes and myelocytes. Conclusion: We identified hematopoietic failure of Rpl11-deficient zebrafish embryos using transcriptome deep sequencing and elucidated potential underlying mechanisms. -

De Novo Frameshift Mutation in ASXL3 in a Patient with Global Developmental Delay, Microcephaly, and Craniofacial Anomalies
Children's Mercy Kansas City SHARE @ Children's Mercy Manuscripts, Articles, Book Chapters and Other Papers 9-17-2013 De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. Darrell L. Dinwiddie Sarah E. Soden Children's Mercy Hospital Carol J. Saunders Children's Mercy Hospital Neil A. Miller Children's Mercy Hospital Emily G. Farrow Children's Mercy Hospital See next page for additional authors Follow this and additional works at: https://scholarlyexchange.childrensmercy.org/papers Part of the Medical Genetics Commons Recommended Citation Dinwiddie, D. L., Soden, S. E., Saunders, C. J., Miller, N. A., Farrow, E. G., Smith, L. D., Kingsmore, S. F. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med Genomics 6, 32-32 (2013). This Article is brought to you for free and open access by SHARE @ Children's Mercy. It has been accepted for inclusion in Manuscripts, Articles, Book Chapters and Other Papers by an authorized administrator of SHARE @ Children's Mercy. For more information, please contact [email protected]. Creator(s) Darrell L. Dinwiddie, Sarah E. Soden, Carol J. Saunders, Neil A. Miller, Emily G. Farrow, Laurie D. Smith, and Stephen F. Kingsmore This article is available at SHARE @ Children's Mercy: https://scholarlyexchange.childrensmercy.org/papers/1414 Dinwiddie et al. BMC Medical Genomics 2013, 6:32 http://www.biomedcentral.com/1755-8794/6/32 CASE REPORT Open Access De novo frameshift -

Metastatic Adrenocortical Carcinoma Displays Higher Mutation Rate and Tumor Heterogeneity Than Primary Tumors
ARTICLE DOI: 10.1038/s41467-018-06366-z OPEN Metastatic adrenocortical carcinoma displays higher mutation rate and tumor heterogeneity than primary tumors Sudheer Kumar Gara1, Justin Lack2, Lisa Zhang1, Emerson Harris1, Margaret Cam2 & Electron Kebebew1,3 Adrenocortical cancer (ACC) is a rare cancer with poor prognosis and high mortality due to metastatic disease. All reported genetic alterations have been in primary ACC, and it is 1234567890():,; unknown if there is molecular heterogeneity in ACC. Here, we report the genetic changes associated with metastatic ACC compared to primary ACCs and tumor heterogeneity. We performed whole-exome sequencing of 33 metastatic tumors. The overall mutation rate (per megabase) in metastatic tumors was 2.8-fold higher than primary ACC tumor samples. We found tumor heterogeneity among different metastatic sites in ACC and discovered recurrent mutations in several novel genes. We observed 37–57% overlap in genes that are mutated among different metastatic sites within the same patient. We also identified new therapeutic targets in recurrent and metastatic ACC not previously described in primary ACCs. 1 Endocrine Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA. 2 Center for Cancer Research, Collaborative Bioinformatics Resource, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA. 3 Department of Surgery and Stanford Cancer Institute, Stanford University, Stanford, CA 94305, USA. Correspondence and requests for materials should be addressed to E.K. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:4172 | DOI: 10.1038/s41467-018-06366-z | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-06366-z drenocortical carcinoma (ACC) is a rare malignancy with types including primary ACC from the TCGA to understand our A0.7–2 cases per million per year1,2. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Whole-Exome Sequencing Associates Novel CSMD1 Gene Mutations with Familial Parkinson Disease
Whole-exome sequencing associates novel CSMD1 gene mutations with familial Parkinson disease Javier Ruiz-Martínez, ABSTRACT MD, PhD Objective: Despite the enormous advancements made in deciphering the genetic architecture of Luis J. Azcona, BBA Parkinson disease (PD), the majority of PD is idiopathic, with single gene mutations explaining only Alberto Bergareche, MD a small proportion of the cases. Jose F. Martí-Massó, MD, Methods: In this study, we clinically evaluated 2 unrelated Spanish families diagnosed with PD, in PhD which known PD genes were previously excluded, and performed whole-exome sequencing anal- Coro Paisán-Ruiz, PhD yses in affected individuals for disease gene identification. Results: Patients were diagnosed with typical PD without relevant distinctive symptoms. Two dif- Correspondence to ferent novel mutations were identified in the CSMD1 gene. The CSMD1 gene, which encodes Dr. Paisán-Ruiz: a complement control protein that is known to participate in the complement activation and [email protected] inflammation in the developing CNS, was previously shown to be associated with the risk of PD in a genome-wide association study. Conclusions: We conclude that the CSMD1 mutations identified in this study might be responsible for the PD phenotype observed in our examined patients. This, along with previous reported studies, may suggest the complement pathway as an important therapeutic target for PD and other neurodegenerative diseases. Neurol Genet 2017;3:e177; doi: 10.1212/NXG.0000000000000177 GLOSSARY AD 5 Alzheimer disease; CCP 5 complement control protein; fPD 5 familial Parkinson disease; H&Y 5 Hoehn and Yahr; INDEL 5 insertions/deletions; LOPD 5 late-onset PD; PD 5 Parkinson disease; RBD 5 REM sleep behavior disorder; RLS 5 restless legs syndrome; SNV 5 single nucleotide variant; WES 5 whole-exome sequencing.