Stabilization of Haloalkane Dehalogenase Structure by Interfacial Interaction with Ionic Liquids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Diversity and Mechanisms of Bacterial Dehalogenation Reactions High Number of Halogen Substituents
O.B. Janssen, T. Bosma and G.J. Poelarends Diversity and Mechanisms of 8acterial Dehalogenation Reactions Abstract Halogenated aliphatic compounds occur widespread as environmental pollutants. Since many of these compounds are xenobiotics and show large dif ferences in degradability which can be correlated to critical steps in catabolic pathways, they are suitable for studies on the evolution of dehalogenating pathways. We have investigated the degradation of 1,2-dichloroethane and 1,3- dichloropropene in detail. For both compounds, the initial step is hydrolytic dehalogenation. The 1,2-dichloroethane and 1,3-dichloropropene dehalogenases we re found to belong to different groups of identical enzymes detected in bac teria isolated from various sites. Genetic analysis and adaptation experiments indicated th at the 1,2-dichloroethane degradation pathway may be of recent evolutionary origin. The large-scale use of 1,3-dichloropropene in agriculture may have contributed to the distribution of genes encoding hydrolytic dehalogenases in the environment. Introduction The biodegradation of synthetic chlorinated chemicals that enter the environ ment is dependent on the capacity of microbial enzymes to recognize these xenobiotic molecules and cleave or labilize carbon-halogen bonds (Janssen et al., 1994). Microbiological studies have led to the isolation of a range of organisms that degrade halogenated aliphatic compounds and use them as a carbon source for growth, and a several dehalogenating enzymes that directly act on carbon halogen bonds have now been identified (Leisinger and Bader 1993; Janssen et al., 1994; Fetzner and Lingens, 1994). In a few cases, the carbon-halogen bond is not directly cleaved but labilized by introduction of other functional groups (Ensley, 1991). -

Classifying Halogenoalkanes Reactions Of
10 Halogenoalkanes H Naming Halogenoalkanes H H H H C H H H H Based on original alkane, with a prefix indicating halogen atom: H C C C H Fluoro for F; Chloro for Cl; Bromo for Br; Iodo for I. H C C C C H Br H H H Cl H H Substituents are listed alphabetically 1-bromopropane 2-chloro-2-methylbutane Classifying Halogenoalkanes Halogenoalkanes can be classified as primary, secondary or tertiary depending on the number of carbon atoms attached to the C-X functional group. H H H H H H H H C H H H H H C C C H H C C C H H C C C C H Br H H H Br H H Cl H H Primary halogenoalkane Secondary halogenoalkane Tertiary halogenoalkane One carbon attached to the Two carbons attached to the Three carbons attached to the carbon atom adjoining the carbon atom adjoining the carbon atom adjoining the halogen halogen halogen Reactions of Halogenoalkanes Halogenoalkanes undergo either substitution or elimination reactions Nucleophilic substitution reactions Substitution: swapping a halogen atom for another atom or groups of atoms - - Nucleophile: electron pair donator e.g. :OH , :NH3, CN :Nu represents any nucleophile – they The Mechanism: We draw (or outline) mechanisms to always have a lone pair and act as show in detail how a reaction proceeds electron pair donators Nu:- The nucleophiles The carbon has a small attack the positive H H H H positive charge because carbon atom of the electronegativity H C C X H C C + X- δ+ δ- Nu difference between the carbon and the halogen H H H H We use curly arrows in mechanisms (with A curly arrow will always two line heads) to show the movement of start from a of electrons or two electrons lone pair the centre of a bond The rate of these substitution reactions depends on the strength Bond enthalpy / of the C-X bond kJmol-1 The weaker the bond, the easier it is to break and the faster the reaction. -

Light-Emitting Dehalogenases: Reconstruction of Multifunctional
Research Article Cite This: ACS Catal. 2019, 9, 4810−4823 pubs.acs.org/acscatalysis Light-Emitting Dehalogenases: Reconstruction of Multifunctional Biocatalysts Radka Chaloupkova,† Veronika Liskova,† Martin Toul,† Klara Markova,† Eva Sebestova,† Lenka Hernychova,‡ Martin Marek,† Gaspar P. Pinto,†,∥ Daniel Pluskal,† Jitka Waterman,§ Zbynek Prokop,†,∥ and Jiri Damborsky*,†,∥ † Loschmidt Laboratories, Department of Experimental Biology and Research Centre for Toxic Compounds in the Environment RECETOX, Masaryk University, Kamenice 5/A13, 625 00 Brno, Czech Republic ‡ Regional Centre for Applied Molecular Oncology, Masaryk Memorial Cancer Institute, 656 53 Brno, Czech Republic § Diamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, United Kingdom ∥ International Clinical Research Center, St. Anne’s University Hospital Brno, Pekarska 53, 656 91 Brno, Czech Republic *S Supporting Information ABSTRACT: To obtain structural insights into the emergence of biological functions from catalytically promiscuous enzymes, we reconstructed an ancestor of catalytically distinct, but evolutionarily related, haloalkane dehalogenases (EC 3.8.1.5) and Renilla luciferase (EC 1.13.12.5). This ancestor has both hydrolase and monooxygenase activities. Its crystal structure solved to 1.39 Å resolution revealed the presence of a catalytic pentad conserved in both dehalogenase and luciferase descend- ants and a molecular oxygen bound in between two residues typically stabilizing a halogen anion. The differences in the conformational dynamics of the specificity-determining cap domains between the ancestral and descendant enzymes were accessed by molecular dynamics and hydrogen−deuterium exchange mass spectrometry. Stopped-flow analysis revealed that the alkyl enzyme intermediate formed in the luciferase-catalyzed reaction is trapped by blockage of a hydrolytic reaction step. -

Biotransformation of Halogenated Compounds by Lyophilized Cells Of
Biotransformation of halogenated compounds by lyophilized cells of Rhodococcus erythropolis in a continuous solid-gas biofilter Benjamin Erable, Thierry Maugard, Isabelle Goubet, Sylvain Lamare, Marie Dominique Legoy To cite this version: Benjamin Erable, Thierry Maugard, Isabelle Goubet, Sylvain Lamare, Marie Dominique Legoy. Biotransformation of halogenated compounds by lyophilized cells of Rhodococcus erythropolis in a continuous solid-gas biofilter. Process Biochemistry, Elsevier, 2005, vol. 40, pp.45-51. 10.1016/j.procbio.2003.11.031. hal-00782060 HAL Id: hal-00782060 https://hal.archives-ouvertes.fr/hal-00782060 Submitted on 29 Jan 2013 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Open Archive Toulouse Archive Ouverte ( OATAO ) OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible. This is an author-deposited version published in: http://oatao.univ-toulouse.fr/ Eprints ID: 7878 To link to this article : DOI:10.1016/j.procbio.2003.11.031 URL: http://dx.doi.org/10.1016/j.procbio.2003.11.031 To cite this version: Erable, Benjamin and Maugard, Thierry and Goubet, Isabelle and Lamare, Sylvain and Legoy, Marie Dominique Biotransformation of halogenated compounds by lyophilized cells of Rhodococcus erythropolis in a continuous solid–gas biofilter. -
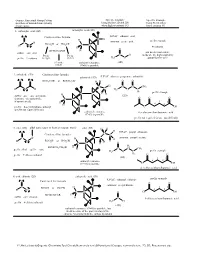
Generic Functional Group Pattern in Ordero of Nomenclature Priority in Our Course. Specific Example Using Suffix (2D and 3D)
Generic Functional Group Pattern Specific Example Specific Example in ordero of nomenclature priority Using Suffix (2D and 3D) Using Prefix when in our course. when highest priority FG lower priority FG 1. carboxylic acid (2D) carboxylic acid (3D) O Condensed line formula IUPAC: ethanoic acid O H H prefix example C H common: acetic acid RCO2H or HO2CR C C R O #-carboxy H O RCH(CO H)R' H suffix: -oic acid 2 O not used in our course FG on FG on C H (acids are the highest priority prefix: #-carboxy the right the left H3C O group that we use) FG in the carbonyl resonance (2D) middle (C=O) is possible 2. anhydride (2D) Condensed line formula anhydride (3D) IUPAC: ethanoic propanoic anhydride O O RCO2COR' or ROCO2CR' H H O O H C C C CH C 3 R O R H3C O C H H 2 prefix example H O C H suffix: -oic -oic anhydride (2D) (just one -oic anhydride, O O O C C O if symmetrical) 3 1 H 4 H 2 prefix: #-acyloxyalkanecarbonyl O O OH (prefix not required for us) carbonyl resonance 4-acyloxymethanebutanoic acid (C=O) is possible (prefix not required for us - too difficult) 3. ester (2D) alkyl name (goes in front as separate word) ester (3D) H H H IUPAC: propyl ethanoate O C Condensed line formula C O C common: propyl acetate C R' H H R O RCO2R' or R'O2CR H H C C H O H H2 RCH(CO2CH3)R' prefix: alkyl suffix: -oate H C C CH3 O H C O C prefix example 3 H prefix: #-alkoxycarbonyl 2 O O (2D) 3 1 carbonyl resonance 4 2 (C=O) is possible O OH 4-methoxycarbonylbutanoic acid 4. -

A Perovskite-Based Paper Microfluidic Sensor for Haloalkane Assays
ORIGINAL RESEARCH published: 26 April 2021 doi: 10.3389/fchem.2021.682006 A Perovskite-Based Paper Microfluidic Sensor for Haloalkane Assays Lili Xie 1, Jie Zan 1, Zhijian Yang 1, Qinxia Wu 1, Xiaofeng Chen 1, Xiangyu Ou 1, Caihou Lin 2*, Qiushui Chen 1,3* and Huanghao Yang 1,3* 1 Ministry of Education (MOE) Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, College of Chemistry, Fuzhou University, Fuzhou, China, 2 Department of Neurosurgery, Fujian Medical University Union Hospital, Fuzhou, China, 3 Fujian Science and Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, China Detection of haloalkanes is of great industrial and scientific importance because some Edited by: haloalkanes are found serious biological and atmospheric issues. The development Jinyi Lin, of a flexible, wearable sensing device for haloalkane assays is highly desired. Here, Nanjing Tech University, China we develop a paper-based microfluidic sensor to achieve low-cost, high-throughput, Reviewed by: Xiaobao Xu, and convenient detection of haloalkanes using perovskite nanocrystals as a nanoprobe Nanjing University of Science and through anion exchanging. We demonstrate that the CsPbX3 (X = Cl, Br, or I) Technology, China nanocrystals are selectively and sensitively in response to haloalkanes (CH2Cl2, CH2Br2), Xuewen Wang, Northwestern Polytechnical and their concentrations can be determined as a function of photoluminescence University, China spectral shifts of perovskite nanocrystals. In particular, an addition of nucleophilic trialkyl *Correspondence: phosphines (TOP) or a UV-photon-induced electron transfer from CsPbX3 nanocrystals is Caihou Lin [email protected] responsible for achieving fast sensing of haloalkanes. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Fully Halogenated Chlorofluorocarbons
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 113 FULLY HALOGENATED CHLOROFLUOROCARBONS This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1990 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination of laboratory testing and epidemiological studies, and promotion of research on the mechanisms of the biological action of chemicals. WHO Library Cataloguing in Publication Data Fully halogenated chlorofluorocarbons. (Environmental health criteria ; 113) 1.Freons - adverse effects 2.Freons - toxicity I.Series ISBN 92 4 157113 6 0 (NLM Classification: QV 633) ISSN 0250-863X The World Health Organization welcomes requests for permission to reproduce or translate its publications, in part or in full. Applications and enquiries should be addressed to the Office of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available. -

Characterization of Hydrolytic Dehalogenases
CHARACTERIZATION OF HYDROLYTIC DEHALOGENASES: SUBSTRATE SPECIFICITY AND CARBON ISOTOPE FRACTIONATION A thesis submitted in conformity with the requirements for the degree of Master of Science. Graduate Department of Cell and Systems Biology University of Toronto © Copyright by Christopher Tran (2013) ABSTRACT: Characterization of Hydrolytic Dehalogenases: Substrate Specificity and Carbon Isotope Fractionation: Christopher Tran, 2013, M.Sc., Department of Cell and Systems Biology, University of Toronto The first project is focused on kinetic analysis of two enzymes: Rsc1362 (Ralstonia solanacearum GMI1000) and PA0810 (Pseudomonas aeruginosa PA01). Rsc1362 had a kcat of 504±66 min-1 and a KM of 0.06±0.02 mM, PA0810 had a kcat of 2.6±0.6 min-1 and a KM of 0.44±0.2 mM. A lack of environmental conteXt for a chloroacetate dehalogenase was noted in Pseudomonas aeruginosa PA01. The second project focuses on kinetic and stable isotope fractionation of 1,2- dichloroethane by DhlA (Xanthobacter autotrophicus GJ10), and Jann2620 (Jannaschia CCS1). Although both enzymes had different kinetics (DhlA: KM = 4.8±0.6 mM and kcat = 133±8 min-1, Jann2620: KM = 25.9±2.3 mM and kcat = ~1.7 min-1), they fractionated similarly (ε values of -33.9‰ and -32.9‰ for DhlA and Jann2620, respectively). As calculated AKIE values were similar to the eXpected values of an abiotic reaction, it was determined that neither enzyme masks the intrinsic fractionation. ii Table of Contents ABSTRACT: ............................................................................................................................... -

Repositioning the Catalytic Triad Aspartic Acid of Haloalkane Dehalogenase: Effects on Stability, Kinetics, and Structure
2 Repositioning the Catalytic Triad Aspartic Acid of Haloalkane Dehalogenase: Effects on Stability, Kinetics, and Structure Geja H. Krooshof, Edwin M. Kwant, Jiří Damborský, Jaroslav Koča, and Dick B. Janssen Biochemistry 36, 9571–9580 (1997) Chapter 2 Repositioning the Catalytic Triad Aspartic Acid of Haloalkane Dehalogenase: Effects on Stability, Kinetics, and Structure Haloalkane dehalogenase (DhlA) catalyzes the hydrolysis of haloalkanes via an alkyl– enzyme intermediate. The covalent intermediate, which is formed by nucleophilic substitution with Asp124, is hydrolyzed by a water molecule that is activated by His289. The role of Asp260, which is the third member of the catalytic triad, was studied by site-directed mutagenesis. Mutation of Asp260 to asparagine resulted in a catalytically inactive D260N mutant, which demonstrates that the triad acid Asp260 is essential for dehalogenase activity. Furthermore, Asp260 has an important structural role, since the D260N enzyme accumulated mainly in inclusion bodies during expression, and neither substrate nor product could bind in the active site cavity. Activity for brominated substrates was restored to D260N by replacing Asn148 with an aspartic or glutamic acid. Both double mutants D260N+N148D and D260N+N148E had a 10-fold reduced kcat and 40-fold higher Km values for 1,2-dibromoethane compared to the wild-type enzyme. Pre-steady-state kinetic analysis of the D260N+N148E double mutant showed that the decrease in kcat was mainly caused by a 220-fold reduction of the rate of carbon–bromine bond cleavage and a 10-fold decrease in the rate of hydrolysis of the alkyl–enzyme intermediate. On the other hand, bromide was released 12-fold faster and via a different pathway than in the wild-type enzyme.