Supplementary Online Materials Title: AI-Guided Discovery of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

E2F1 Interactive with BRCA1 Pathway Induces HCC Two Different Small Molecule Metabolism Or Cell Cycle Regulation Via Mitochondrion Or CD4+T to Cytosol
Received: 29 March 2017 | Accepted: 3 May 2017 DOI: 10.1002/jcp.25988 ORIGINAL RESEARCH ARTICLE E2F1 interactive with BRCA1 pathway induces HCC two different small molecule metabolism or cell cycle regulation via mitochondrion or CD4+T to cytosol Qingchun Chen1 | Lin Wang1 | Minghu Jiang2 | Juxiang Huang1 | Zhenfu Jiang1 | Haitao Feng3 | Zhili Ji4 1 Computation and Systems Biology, School of Electronic Engineering, Beijing University of Breast cancer 1 (BRCA1) and E2F transcription factor 1 (E2F1) are related to metabolism Posts and Telecommunications, Beijing, China and cell cycle regulation. However, the corresponding mechanism is not clear in HCC. 2 Lab of Computational Linguistics, School of Humanities and Social Sciences, Tsinghua High BRCA1 direct pathway was constructed with 11 molecules from E2F1 feedback- University, Beijing, China interactive network in HCC by GRNInfer based on 39 Pearson mutual positive 3 Dean department, Heilongjiang University of corelation CC ≥0.25 molecules with E2F1. Integration of GRNInfer with GO, KEGG, Chinese Medicine, Harbin, China BioCarta, GNF_U133A, UNIGENE_EST, Disease, GenMAPP databases by DAVID and 4 Department of General Surgery, Beijing Luhe Hospital, Capital Medical University, Beijing, China MAS 3.0, E2F1 feedback-interactive BRCA1 indirect mitochondrion to cytosol pathway was identified as upstream LAPTM4B activation, feedback UNG, downstream BCAT1- Correspondence Prof., Dr. Lin Wang, Computation and HIST1H2AD-TK1 reflecting protein, and DNA binding with enrichment of small molecule Systems Biology, School of Electronics metabolism; The corresponding BRCA1 indirect membrane to cytosol pathway as Engineering, Beijing University of Posts and Telecommunications, Beijing, 100876, China. upstream CCNB2-NUSAP1 activation, feedback TTK-HIST1H2BJ-CENPF, downstream (Minghu Jiang c/o Lin Wang). -

Supporting Information
Supporting Information Edgar et al. 10.1073/pnas.1601895113 SI Methods (Actimetrics), and recordings were analyzed using LumiCycle Mice. Sample size was determined using the resource equation: Data Analysis software (Actimetrics). E (degrees of freedom in ANOVA) = (total number of exper- – Cell Cycle Analysis of Confluent Cell Monolayers. NIH 3T3, primary imental animals) (number of experimental groups), with −/− sample size adhering to the condition 10 < E < 20. For com- WT, and Bmal1 fibroblasts were sequentially transduced − − parison of MuHV-4 and HSV-1 infection in WT vs. Bmal1 / with lentiviral fluorescent ubiquitin-based cell cycle indicators mice at ZT7 (Fig. 2), the investigator did not know the genotype (FUCCI) mCherry::Cdt1 and amCyan::Geminin reporters (32). of the animals when conducting infections, bioluminescence Dual reporter-positive cells were selected by FACS (Influx Cell imaging, and quantification. For bioluminescence imaging, Sorter; BD Biosciences) and seeded onto 35-mm dishes for mice were injected intraperitoneally with endotoxin-free lucif- subsequent analysis. To confirm that expression of mCherry:: Cdt1 and amCyan::Geminin correspond to G1 (2n DNA con- erin (Promega E6552) using 2 mg total per mouse. Following < ≤ anesthesia with isofluorane, they were scanned with an IVIS tent) and S/G2 (2 n 4 DNA content) cell cycle phases, Lumina (Caliper Life Sciences), 15 min after luciferin admin- respectively, cells were stained with DNA dye DRAQ5 (abcam) and analyzed by flow cytometry (LSR-Fortessa; BD Biosci- istration. Signal intensity was quantified using Living Image ences). To examine dynamics of replicative activity under ex- software (Caliper Life Sciences), obtaining maximum radiance perimental confluent conditions, synchronized FUCCI reporter for designated regions of interest (photons per second per − − − monolayers were observed by time-lapse live cell imaging over square centimeter per Steradian: photons·s 1·cm 2·sr 1), relative 3 d (Nikon Eclipse Ti-E inverted epifluorescent microscope). -

Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data
Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data Hai-Son Phuoc Le May 2013 CMU-ML-13-101 Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data Hai-Son Phuoc Le May 2013 CMU-ML-13-101 Machine Learning Department School of Computer Science Carnegie Mellon University Thesis Committee Ziv Bar-Joseph, Chair Christopher Langmead Roni Rosenfeld Quaid Morris Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy. Copyright @ 2013 Hai-Son Le This research was sponsored by the National Institutes of Health under grant numbers 5U01HL108642 and 1R01GM085022, the National Science Foundation under grant num- bers DBI0448453 and DBI0965316, and the Pittsburgh Life Sciences Greenhouse. The views and conclusions contained in this document are those of the author and should not be interpreted as representing the official policies, either expressed or implied, of any sponsoring institution, the U.S. government or any other entity. Keywords: genomics, gene expression, gene regulation, microarray, RNA-Seq, transcriptomics, error correction, comparative genomics, regulatory networks, cross-species, expression database, Gene Expression Omnibus, GEO, orthologs, microRNA, target prediction, Dirichlet Process, Indian Buffet Process, hidden Markov model, immune response, cancer. To Mom and Dad. i Abstract Advances in genomics allow researchers to measure the complete set of transcripts in cells. These transcripts include messenger RNAs (which encode for proteins) and microRNAs, short RNAs that play an important regulatory role in cellular networks. While this data is a great resource for reconstructing the activity of networks in cells, it also presents several computational challenges. These challenges include the data collection stage which often results in incomplete and noisy measurement, developing methods to integrate several experiments within and across species, and designing methods that can use this data to map the interactions and networks that are activated in specific conditions. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -

Mai Muudatuntuu Ti on Man Mini
MAIMUUDATUNTUU US009809854B2 TI ON MAN MINI (12 ) United States Patent ( 10 ) Patent No. : US 9 ,809 ,854 B2 Crow et al. (45 ) Date of Patent : Nov . 7 , 2017 Whitehead et al. (2005 ) Variation in tissue - specific gene expression ( 54 ) BIOMARKERS FOR DISEASE ACTIVITY among natural populations. Genome Biology, 6 :R13 . * AND CLINICAL MANIFESTATIONS Villanueva et al. ( 2011 ) Netting Neutrophils Induce Endothelial SYSTEMIC LUPUS ERYTHEMATOSUS Damage , Infiltrate Tissues, and Expose Immunostimulatory Mol ecules in Systemic Lupus Erythematosus . The Journal of Immunol @(71 ) Applicant: NEW YORK SOCIETY FOR THE ogy , 187 : 538 - 552 . * RUPTURED AND CRIPPLED Bijl et al. (2001 ) Fas expression on peripheral blood lymphocytes in MAINTAINING THE HOSPITAL , systemic lupus erythematosus ( SLE ) : relation to lymphocyte acti vation and disease activity . Lupus, 10 :866 - 872 . * New York , NY (US ) Crow et al . (2003 ) Microarray analysis of gene expression in lupus. Arthritis Research and Therapy , 5 :279 - 287 . * @(72 ) Inventors : Mary K . Crow , New York , NY (US ) ; Baechler et al . ( 2003 ) Interferon - inducible gene expression signa Mikhail Olferiev , Mount Kisco , NY ture in peripheral blood cells of patients with severe lupus . PNAS , (US ) 100 ( 5 ) : 2610 - 2615. * GeneCards database entry for IFIT3 ( obtained from < http : / /www . ( 73 ) Assignee : NEW YORK SOCIETY FOR THE genecards. org /cgi - bin / carddisp .pl ? gene = IFIT3 > on May 26 , 2016 , RUPTURED AND CRIPPLED 15 pages ) . * Navarra et al. (2011 ) Efficacy and safety of belimumab in patients MAINTAINING THE HOSPITAL with active systemic lupus erythematosus : a randomised , placebo FOR SPECIAL SURGERY , New controlled , phase 3 trial . The Lancet , 377 :721 - 731. * York , NY (US ) Abramson et al . ( 1983 ) Arthritis Rheum . -

Identification of De Novo Mutations in Prenatal Neurodevelopment-Associated Genes In
medRxiv preprint doi: https://doi.org/10.1101/19011007; this version posted November 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of de novo mutations in prenatal neurodevelopment-associated genes in schizophrenia in two Han Chinese patient-sibling family-based cohorts Running head: Identification of de novo mutations in schizophrenia in Chinese families Shan Jiang1,*, Daizhan Zhou2,3,*, Yin-Ying Wang1, Peilin jia1, Chunling Wan2,3, Xingwang Li2,3, Guang He2,3, Dongmei Cao2, Xiaoqian Jiang4, Kenneth S. Kendler5, Ming Tsuang6, Travis Mize7,8, Jain-Shing Wu7, Yimei Lu7, Lin He2,3,9,#, Jingchun Chen7,#, Zhongming Zhao1,10,11,# and Xiangning Chen12,# 1Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA. 2Bio-X Institutes, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Collaborative Innovation Center for Brain Science, Shanghai Jiao Tong University, Shanghai, China. 3Shanghai Key Laboratory of Psychotic Disorders, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 4School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA. 5Virginia Institute of Psychiatric and Behavioral Genetics, Medical College of Virginia and Virginia Commonwealth University, Richmond, VA, 23298, USA. 6Department of Psychiatry, University of California at San Diego, San Diego, CA, 92093, USA. 7Nevada Institute of Personalized Medicine, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. -

Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience-Dependent Cortical Plasticity
New Research Development Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience- Dependent Cortical Plasticity Milo R. Smith1,2,3,4,5,6,7,8, Poromendro Burman1,3,4,5,8, Masato Sadahiro1,3,4,5,6,8, Brian A. Kidd,2,7 Joel T. Dudley,2,7 and Hirofumi Morishita1,3,4,5,8 DOI:http://dx.doi.org/10.1523/ENEURO.0240-16.2016 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 2Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 3Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 4Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 5Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 6Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 7Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, and 8Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029 Visual Abstract Throughout childhood and adolescence, periods of heightened neuroplasticity are critical for the development of healthy brain function and behavior. Given the high prevalence of neurodevelopmental disorders, such as autism, identifying disruptors of developmental plasticity represents an essential step for developing strategies for prevention and intervention. Applying a novel computational approach that systematically assessed connections between 436 transcriptional signatures of disease and multiple signatures of neuroplasticity, we identified inflammation as a common pathological process central to a diverse set of diseases predicted to dysregulate Significance Statement During childhood and adolescence, heightened neuroplasticity allows the brain to reorganize and adapt to its environment. -

Supplemental Data.Pdf
Supplementary material -Table of content Supplementary Figures (Fig 1- Fig 6) Supplementary Tables (1-13) Lists of genes belonging to distinct biological processes identified by GREAT analyses to be significantly enriched with UBTF1/2-bound genes Supplementary Table 14 List of the common UBTF1/2 bound genes within +/- 2kb of their TSSs in NIH3T3 and HMECs. Supplementary Table 15 List of gene identified by microarray expression analysis to be differentially regulated following UBTF1/2 knockdown by siRNA Supplementary Table 16 List of UBTF1/2 binding regions overlapping with histone genes in NIH3T3 cells Supplementary Table 17 List of UBTF1/2 binding regions overlapping with histone genes in HMEC Supplementary Table 18 Sequences of short interfering RNA oligonucleotides Supplementary Table 19 qPCR primer sequences for qChIP experiments Supplementary Table 20 qPCR primer sequences for reverse transcription-qPCR Supplementary Table 21 Sequences of primers used in CHART-PCR Supplementary Methods Supplementary Fig 1. (A) ChIP-seq analysis of UBTF1/2 and Pol I (POLR1A) binding across mouse rDNA. UBTF1/2 is enriched at the enhancer and promoter regions and along the entire transcribed portions of rDNA with little if any enrichment in the intergenic spacer (IGS), which separates the rDNA repeats. This enrichment coincides with the distribution of the largest subunit of Pol I (POLR1A) across the rDNA. All sequencing reads were mapped to the published complete sequence of the mouse rDNA repeat (Gene bank accession number: BK000964). The graph represents the frequency of ribosomal sequences enriched in UBTF1/2 and Pol I-ChIPed DNA expressed as fold change over those of input genomic DNA. -

Investigating the Effect of Chronic Activation of AMP-Activated Protein
Investigating the effect of chronic activation of AMP-activated protein kinase in vivo Alice Pollard CASE Studentship Award A thesis submitted to Imperial College London for the degree of Doctor of Philosophy September 2017 Cellular Stress Group Medical Research Council London Institute of Medical Sciences Imperial College London 1 Declaration I declare that the work presented in this thesis is my own, and that where information has been derived from the published or unpublished work of others it has been acknowledged in the text and in the list of references. This work has not been submitted to any other university or institute of tertiary education in any form. Alice Pollard The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives license. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the license terms of this work. 2 Abstract The prevalence of obesity and associated diseases has increased significantly in the last decade, and is now a major public health concern. It is a significant risk factor for many diseases, including cardiovascular disease (CVD) and type 2 diabetes. Characterised by excess lipid accumulation in the white adipose tissue, which drives many associated pathologies, obesity is caused by chronic, whole-organism energy imbalance; when caloric intake exceeds energy expenditure. Whilst lifestyle changes remain the most effective treatment for obesity and the associated metabolic syndrome, incidence continues to rise, particularly amongst children, placing significant strain on healthcare systems, as well as financial burden. -
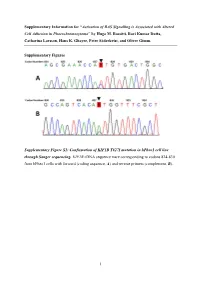
Activation of RAS Signalling Is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M
Supplementary Information for “Activation of RAS Signalling is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M. Rossitti, Ravi Kumar Dutta, Catharina Larsson, Hans K. Ghayee, Peter Söderkvist, and Oliver Gimm. Supplementary Figures Supplementary Figure S1: Confirmation of KIF1B T827I mutation in hPheo1 cell line through Sanger sequencing. KIF1B cDNA sequence trace corresponding to codons 824-830 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 1 Supplementary Figure S2: Confirmation of NRAS Q61K mutation in hPheo1 cell line through Sanger sequencing. NRAS cDNA sequence trace corresponding to codons 56-67 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 2 Supplementary Figure S3: CCND1 gene expression and hPheo1 proliferation. A: Expression of CCND1 mRNA assessed by RT-qPCR and presented as fold change (2-ΔΔCT, mean ± standard error of the mean). B: Cell counts at 1, 2, and 3 days after plating (corresponding to 72, 96 and 120 hours posttransfection, respectively) of control- or siNRAS#1-transfected hPheo1 cells expressed as fold change of the number of cells plated at day 0 (48 hours posttransfection; mean ± standard deviation). All results are from three independent siRNA experiments. 3 Supplementary Tables Supplementary Table S1: List of transcript cluster IDs significantly upregulated in hPheo1 by siNRAS treatment (comparison: siNRAS versus control-transfected hPheo1; ANOVA p < 0.05, FDR < 0.25, fold change < -1.5 or > 1.5). Transcript -

Whole-Genome Sequencing Identifies New Genetic Alterations in Meningiomas
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 10), pp: 17070-17080 Research Paper Whole-genome sequencing identifies new genetic alterations in meningiomas Mei Tang1,*, Heng Wei2,*, Lu Han1,*, Jiaojiao Deng3, Yuelong Wang3, Meijia Yang1, Yani Tang1, Gang Guo1, Liangxue Zhou3, Aiping Tong1 1The State Key Laboratory of Biotherapy and Cancer Center/Collaborative Innovation Center of Biotherapy, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China 2College of Life Science, Sichuan University, Chengdu 610064, China 3Department of Neurosurgery, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China *These authors have contributed equally to the work Correspondence to: Aiping Tong, email: [email protected] Liangxue Zhou, email: [email protected] Keywords: whole-genome sequencing, meningioma, chromosome instability, copy number alteration, mutation Received: October 24, 2016 Accepted: January 13, 2017 Published: February 03, 2017 ABSTRACT The major known genetic contributor to meningioma formation was NF2, which is disrupted by mutation or loss in about 50% of tumors. Besides NF2, several recurrent driver mutations were recently uncovered through next-generation sequencing. Here, we performed whole-genome sequencing across 7 tumor-normal pairs to identify somatic genetic alterations in meningioma. As a result, Chromatin regulators, including multiple histone members, histone-modifying enzymes and several epigenetic regulators, are the major category among all of the identified copy number variants and single nucleotide variants. Notably, all samples contained copy number variants in histone members. Recurrent chromosomal rearrangements were detected on chromosome 22q, 6p21-p22 and 1q21, and most of the histone copy number variants occurred in these regions. -

BMC Genetics Biomed Central
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector BMC Genetics BioMed Central Research article Open Access A novel replication-independent histone H2a gene in mouse Hiromi Nishida*, Takahiro Suzuki, Yasuhiro Tomaru and Yoshihide Hayashizaki Address: Laboratory for Genome Exploration Research Group, RIKEN Genomic Sciences Center (GSC), RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan Email: Hiromi Nishida* - [email protected]; Takahiro Suzuki - [email protected]; Yasuhiro Tomaru - [email protected]; Yoshihide Hayashizaki - [email protected] * Corresponding author Published: 19 February 2005 Received: 07 October 2004 Accepted: 19 February 2005 BMC Genetics 2005, 6:10 doi:10.1186/1471-2156-6-10 This article is available from: http://www.biomedcentral.com/1471-2156/6/10 © 2005 Nishida et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: An uncharacterized histone H2a-coding transcript (E130307C13) has been cloned from a mouse full-length cDNA library. This transcript is encoded on chromosome 6, approximately 4 kb upstream of a histone H4 gene, Hist4h4. The proteins encoded by this transcript and the human H2afj mRNA isoform-2 have the highest amino acid similarity. In this paper, we characterize it from the expression pattern given by quantitative RT-PCR. Results: Quantitative RT-PCR indicated that the gene that encodes E130307C13 (E130307C13) is regulated in a replication-independent manner, and therefore it is H2afj.