LECTURE 6 Electrophilic Addition to Alkynes Alkynes Are Very Similar To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

United States Patent (19) 11) 4,219,685 Savini 45 Aug
United States Patent (19) 11) 4,219,685 Savini 45 Aug. 26, 1980 (54) IMPROVING ODOR OF ISOPROPANOL Attorney, Agent, or Firm-C. Leon Kim; Rebecca Yablonsky 75 Inventor: Charles Savini, Warren, N.J. 57 ABSTRACT 73 Assignee: Exxon Research & Engineering Co., Methods for deodorizing lower alcohols such as ethanol Florham Park, N.J. and isopropyl alcohol and their oxy derivatives such as ethers and esters are disclosed, including contacting 21 Appl. No.: 31,761 these compounds with a deodorizing contact mass com 22 Filed: Apr. 20, 1979 prising, on a support, metals and/or metal oxides, pref erably of the metals of Group IB, VIB and VIII of the Periodic Table, where the metal oxides are at least par Related U.S. Application Data tially reduced to metal and the deodorizing contact 63 Continuation-in-part of Ser. No. 908,910, May 24, mass has a minimum particle dimension of greater than 1978, abandoned, which is a continuation-in-part of about 0.254 mm so as to be in a form suitable for use in Ser. No. 834,240, Sep. 19, 1977, abandoned. a fixed bed contacting process. In a preferred embodi ment, isopropyl alcohol is deodorized employing such 51 Int. Cl’.............................................. CO7C 29/24 deodorizing contact masses, preferably comprising 52 U.S. Cl. .............. ... 568/917; 568/922 Group VIII metals such as nickel, iron, cobalt and the 58) Field of Search ................................ 568/917,922 like, on a support, so that the contact mass has a surface 56) References Cited area less than about 1,500 m2 per gram. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Mass Transfer and Hydrogenation of Acetone
MASS TRANSFER AND HYDROGENATION OF ACETONE IN A VIBRATING SLURRY REACTOR Thesis submitted for the degree of Ph.D. of the University of London by Norberto Oscar Lemcoff Department-of Chemical Engineering and Chemical Technology, Imperial College of Science and Technology, London S.W.7. February, 1974 ABSTRACT When a column of liquid-is made to oscillate vertically, gas bubbles are entrained and carried to the bottom of the column, where they aggregate and form a large slug which, in turn, rises to the top. Large volumes of gas are entrained and the fluid becomes highly agitated. A study of the solid- liquid mass transfer and of an heterogeneous catalyzed reaction in this equipment and the kinetic analysis of the hydrogenation of acetone over Raney nickel have been carried out. A large increase in the mass transfer coefficient for solid-liquid systems in the vibrating liquid column over those reported in a stirred tank has been observed. Two correlations for the Sherwood number as a function of the Reynolds, Schmidt and Froude numbers and the relative amplitude of oscillation have been found, one corresponding to the case no bubble cycling occurs and the other to the case it does. In the kinetic study, a Langmuir-Hinshelwood type equa- tion to represent the rate of hydrogenation of acetone in n-octane, isooctane, isopropanol and water and a correlation for the hydrogen solubility in different solvents and their mixtures have been developed. Activation energies and the order of reaction with respect to hydrogen have been determined. Finally, the behaviour of the vibrating column of liquid as an heterogeneous slurry reactor has been analysed by using two Raney nickel catalysts of different average particle size in the hydrogenation of aqueous acetone. -

Transfer Hydrogenation of Olefins Catalysed by Nickel Nanoparticles
View metadata, citation and similar papers at core.ac.uk brought to you by CORE ARTICLE IN PRESS TET19911_proofprovided by Repositorio 26 October Institucional 2009 de la Universidad 1/7 de Alicante Tetrahedron xxx (2009) 1–7 Contents lists available at ScienceDirect Tetrahedron journal homepage: www.elsevier.com/locate/tet 55 1 56 2 Transfer hydrogenation of olefins catalysed by nickel nanoparticles 57 3 58 4 59 ** * 5 Francisco Alonso , Paola Riente, Miguel Yus 60 6 Departamento de Quı´mica Orga´nica, Facultad de Ciencias and Instituto de Sı´ntesis Orga´nica (ISO), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain 61 7 62 8 63 9 article info abstract 64 10 65 11 Article history: Nickel nanoparticles have been found to effectively catalyse the hydrogen-transfer reduction of a variety 66 12 Received 22 September 2009 of non-functionalised and functionalised olefins using 2-propanol as the hydrogen donor. The hetero- 67 13 Received in revised form geneous process has been shown to be highly chemoselective for certain substrates, with all the cor- 68 14 October 2009 responding alkanes being obtained in high yields. A synthesis of the natural dihydrostilbene brittonin 69 14 Accepted 15 October 2009 A is also reported based on the use of nickel nanoparticles. 15 Available online xxx 70 16 Ó 2009 Published by Elsevier Ltd. 71 17 Keywords: PROOF 72 18 Hydrogen transfer 73 19 Reduction 74 Olefins 75 20 Nickel nanoparticles 21 76 22 77 23 78 24 79 25 1. Introduction of chiral ligands. In contrast with the reduction of carbonyl com- 80 26 pounds, the hydrogen-transfer reduction of olefins has been little 81 27 The reduction of carbon–carbon double bonds is one of the studied, mainly involving noble-metal catalysts. -

Effect of Preparation Conditions of Raney Nickel on Its Catalytic Properties for Slurry Phase Hydrogenation of O-Nitrophenol to O-Aminophenol
Indian Journal of Chemical Technology Vol. 5, July 1998, pp. 199-208 Effect of preparation conditions of Raney Nickel on its catalytic properties for slurry phase hydrogenation of o-nitrophenol to o-aminophenol V R Choudhary'& M G Sane Chemical Engineering Division, National Chemical Laboratory, Pune 411 008, India Received 5 November 1997; accepted 10 June 1998 Influence of leaching conditions of Ni-Al alloy (50% AI) on the catalytic activity of Raney Nt (m the hydrogenation of o-nitrophenol at 308 K and H2-pressure of 1508 kPa) has been studied and optimum conditions for the preparation of Raney-Ni catalyst for the hydrogenation process have been obtained. The hydrogenation activity of Raney-Ni is found to be strongly influenced by its preparation conditions. Catalytic slurry phase hydrogenation of 0• nitrotoluene over Raney-Ni prepared under nitrophenol using Raney Ni catalyst is an different conditions4 has indicated a strong important commercial process for the production influence of leaching conditions of Ni-Al (50%, AI) of o-aminophenol, an important intermediate for alloy (viz. type of alkali, concentration of alkali, drugs, dyes and pesticides. Nickel aluminium alloy temperature and duration of leaching, and the is commonly used for the preparation of Raney• washing agent (viz. tap water, distilled water, nickel. The alloy contains NiAI3, Ni2A13, and Ni3AI deionised distilled water, 50% ethanol and 95% phases 1.2 and the composition of these phases ethanol) used in the washing of the leached alloy depends on the alloy composition. The non• on the hydrogen content and hydrogenation catalytic part of the alloy (i.e. -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
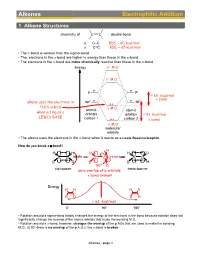
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation. -

Subject Index
455 Subject Index Aminohydroxylation, 364 a-Aminoketone, 281 4-Aminophenol, 18 A Aminothiophene, 158 Abnormal Claisen rearrangement, 1 a-Aminothiophenols, 184 Acrolein, 378 Ammonium ylide, 383 2-(Acylamino)-toluenes, 245 Angeli-Rimini hydroxamic acid Acylation, 100, 145, 200 synthesis, 9 Acyl azides, 98 ~-Anomer, 225 Acylium ion, 145, 149, 175, 177 Anomeric center, 211 a-Acyloxycarboxamides, 298 Anomeric effect, 135 a-Acyloxyketones, 17 ANRORC mechanism, 10 a-Acyloxythioethers, 327 Anthracenes, 51 Acyl transfer, 17, 42, 228, 298, 305, Anti-Markovnikov addition, 219 327,345 Amdt-Eistert homologation, 11 AIBN, 22, 23, 415 Aryl-acetylene, 66 Alder ene reaction, 2 Arylation, 253 Alder's endo rule, Ill 0-Aryliminoethers, 67 Aldol condensation, 3, 14, 26, 34, 2-Arylindoles, 38 69, 130, 147, 172, 305, Aryl migration, 31 340,396,412 Autoxidation, 69, 115, 118 Aldosylamine, 8 Auwers reaction, 13 Alkyl migration, 16, 132, 315, 443 Axial, 347 Alkylation, 144, 145 Azalactone, I 00 N-Aikylation, 162 Azides, 125, 330 Alkylidene carbene, 151 Azirine, 6, 7, 281 Allan-Robinson reaction, 4, 228 Azulene, 310 Allene, 119 1t-Allyl complex, 414 B Allylation, 213, 414 Baeyer-Drewson Allylstannane, 213 indigo synthesis, 14 Allylsilanes, 349 Baeyer-Villiger oxidation, 16, 53 Alper carbonylation, 6 Baker-Venkataraman Alpine-borane®, 262 rearrangement, 17 Aluminum phenolate, 149 Balz-Schiemann reaction, 354 Amadori rearrangement, 8 Bamberger rearrangement, 18 Amide acetal, 74 Bamford-Stevens reaction, 19 Amides, 28, 67,276,339, 356 Bargellini reaction, 20 Amidine, -

Organic Seminar Abstracts
L I B RA R.Y OF THE UN IVE.R.SITY Of ILLI NOIS 547 l£6s \ 954/55 PV Return this book on or before the Latest Date stamped below. University of Illinois Library Llf.1—H41 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/organicsemi195455univ \7^ — SEMINAR TOPICS CHEMISTRY 435 I Semester 1954-55 The Quinolizinium Ion and Some of its Derivatives Harvey M. Loux, September 24 1 The Stereochemistry of Atropine and Cocaine Ho E . Knipmeyer, September 24 4 Recent Developments in the Chemistry of Cinnoline Derivatives Roger H. Kottke , October 1 8 Interpretation of Electrophiiic Aromatic Substitution and Solvolysis of Allylic and Benzhydryl Chlorides in Terms of Hyperconjugation Robert D. Stolow, October 1 11 The Decahydronaphtholic Acids and Their Relationship to the Decalols and the Decalylamines Robert J. Harder, October 8 14 Bis-Cyclopentadienyl Metal Compounds Edwin L. DeYoung, October 8 17 Ion-Pairs as Intermediates in Solvolysis Reactions Arthur H. Goldkamp, October 15 21 Synthesis of Morphine Mohan D. Nair, October 15 24 Structural and Geometrical Isomerism in the Oxidation of Azo Compounds D. F. Morrow, October 22 27 Stereochemical Aspects of Thermochromism John W. Johnson, Jr., October 22 30 A New Method for the Preparation of Olefins --The Pyrolysis of Sulfites F. M. Scheidt, October 29 33 Recent Studies of Macrocyclic Ring Systems: Cyclophanes Fred P. Hauck, Jr., October 29 36 Mechanism of the Para-Claisen Rearrangement Hugh H . Gibbs , November 5 40 A Proposed Mechanism for Basic c is -Dehydrohalogenat ion Robert M. -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary.