Related Plasticity in Classical Trigeminal Neuralgia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Lecture 12 Notes
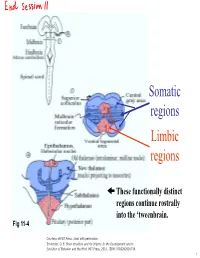
Somatic regions Limbic regions These functionally distinct regions continue rostrally into the ‘tweenbrain. Fig 11-4 Courtesy of MIT Press. Used with permission. Schneider, G. E. Brain structure and its Origins: In the Development and in Evolution of Behavior and the Mind. MIT Press, 2014. ISBN: 9780262026734. 1 Chapter 11, questions about the somatic regions: 4) There are motor neurons located in the midbrain. What movements do those motor neurons control? (These direct outputs of the midbrain are not a subject of much discussion in the chapter.) 5) At the base of the midbrain (ventral side) one finds a fiber bundle that shows great differences in relative size in different species. Give examples. What are the fibers called and where do they originate? 8) A decussating group of axons called the brachium conjunctivum also varies greatly in size in different species. It is largest in species with the largest neocortex but does not come from the neocortex. From which structure does it come? Where does it terminate? (Try to guess before you look it up.) 2 Motor neurons of the midbrain that control somatic muscles: the oculomotor nuclei of cranial nerves III and IV. At this level, the oculomotor nucleus of nerve III is present. Fibers from retina to Superior Colliculus Brachium of Inferior Colliculus (auditory pathway to thalamus, also to SC) Oculomotor nucleus Spinothalamic tract (somatosensory; some fibers terminate in SC) Medial lemniscus Cerebral peduncle: contains Red corticospinal + corticopontine fibers, + cortex to hindbrain fibers nucleus (n. ruber) Tectospinal tract Rubrospinal tract Courtesy of MIT Press. Used with permission. Schneider, G. -

Brainstem and Its Associated Cranial Nerves
Brainstem and its Associated Cranial Nerves Anatomical and Physiological Review By Sara Alenezy With appreciation to Noura AlTawil’s significant efforts Midbrain (Mesencephalon) External Anatomy of Midbrain 1. Crus Cerebri (Also known as Basis Pedunculi or Cerebral Peduncles): Large column of descending “Upper Motor Neuron” fibers that is responsible for movement coordination, which are: a. Frontopontine fibers b. Corticospinal fibers Ventral Surface c. Corticobulbar fibers d. Temporo-pontine fibers 2. Interpeduncular Fossa: Separates the Crus Cerebri from the middle. 3. Nerve: 3rd Cranial Nerve (Oculomotor) emerges from the Interpeduncular fossa. 1. Superior Colliculus: Involved with visual reflexes. Dorsal Surface 2. Inferior Colliculus: Involved with auditory reflexes. 3. Nerve: 4th Cranial Nerve (Trochlear) emerges caudally to the Inferior Colliculus after decussating in the superior medullary velum. Internal Anatomy of Midbrain 1. Superior Colliculus: Nucleus of grey matter that is associated with the Tectospinal Tract (descending) and the Spinotectal Tract (ascending). a. Tectospinal Pathway: turning the head, neck and eyeballs in response to a visual stimuli.1 Level of b. Spinotectal Pathway: turning the head, neck and eyeballs in response to a cutaneous stimuli.2 Superior 2. Oculomotor Nucleus: Situated in the periaqueductal grey matter. Colliculus 3. Red Nucleus: Red mass3 of grey matter situated centrally in the Tegmentum. Involved in motor control (Rubrospinal Tract). 1. Inferior Colliculus: Nucleus of grey matter that is associated with the Tectospinal Tract (descending) and the Spinotectal Tract (ascending). Tectospinal Pathway: turning the head, neck and eyeballs in response to a auditory stimuli. 2. Trochlear Nucleus: Situated in the periaqueductal grey matter. Level of Inferior 3. -

Somatotopic Organization of Perioral Musculature Innervation Within the Pig Facial Motor Nucleus
Original Paper Brain Behav Evol 2005;66:22–34 Received: September 20, 2004 Returned for revision: November 10, 2004 DOI: 10.1159/000085045 Accepted after revision: December 7, 2004 Published online: April 8, 2005 Somatotopic Organization of Perioral Musculature Innervation within the Pig Facial Motor Nucleus Christopher D. Marshall a Ron H. Hsu b Susan W. Herring c aTexas A&M University at Galveston, Galveston, Tex., bDepartment of Pediatric Dentistry, University of North Carolina, Chapel Hill, N.C., and cDepartment of Orthodontics, University of Washington, Seattle, Wash., USA Key Words pools of the lateral 4 of the 7 subnuclei of the facial motor Somatotopy W Innervation W Facial nucleus W Perioral nucleus. The motor neuron pools of the perioral muscles muscles W Orbicularis oris W Buccinator W Mammals were generally segregated from motoneurons innervat- ing other facial muscles of the rostrum. However, motor neuron pools were not confined to single nuclei but Abstract instead spanned across 3–4 subnuclei. Perioral muscle The orbicularis oris and buccinator muscles of mammals motor neuron pools overlapped but were organized so- form an important subset of the facial musculature, the matotopically. Motor neuron pools of portions of the perioral muscles. In many taxa, these muscles form a SOO overlapped greatly with each other but exhibited a robust muscular hydrostat capable of highly manipula- crude somatotopy within the SOO motor neuron pool. tive fine motor movements, likely accompanied by a spe- The large and somatotopically organized SOO motor cialized pattern of innervation. We conducted a retro- neuron pool in pigs suggests that the upper lip might be grade nerve-tracing study of cranial nerve (CN) VII in pigs more richly innervated than the other perioral muscles (Sus scrofa) to: (1) map the motor neuron pool distribu- and functionally divided. -

A Systematic Review of Structural and Functional MRI Stud
Yin et al. The Journal of Headache and Pain (2020) 21:78 The Journal of Headache https://doi.org/10.1186/s10194-020-01131-4 and Pain REVIEW ARTICLE Open Access The neuro-pathophysiology of temporomandibular disorders-related pain: a systematic review of structural and functional MRI studies Yuanyuan Yin1,2†, Shushu He2†, Jingchen Xu2, Wanfang You1,3, Qian Li1,3, Jingyi Long1,3, Lekai Luo1,3, Graham J. Kemp4, John A. Sweeney1,5, Fei Li1,3*, Song Chen2* and Qiyong Gong1,3 Abstract Chronic pain surrounding the temporomandibular joints and masticatory muscles is often the primary chief complaint of patients with temporomandibular disorders (TMD) seeking treatment. Yet, the neuro-pathophysiological basis underlying it remains to be clarified. Neuroimaging techniques have provided a deeper understanding of what happens to brain structure and function in TMD patients with chronic pain. Therefore, we performed a systematic review of magnetic resonance imaging (MRI) studies investigating structural and functional brain alterations in TMD patients to further unravel the neurobiological underpinnings of TMD-related pain. Online databases (PubMed, EMBASE, and Web of Science) were searched up to August 3, 2019, as complemented by a hand search in reference lists. A total of 622 papers were initially identified after duplicates removed and 25 studies met inclusion criteria for this review. Notably, the variations of MRI techniques used and study design among included studies preclude a meta-analysis and we discussed the findings qualitatively according to the specific neural system or network the brain regions were involved in. Brain changes were found in pathways responsible for abnormal pain perception, including the classic trigemino-thalamo-cortical system and the lateral and medial pain systems. -

MR Imaging of Wallerian Degeneration in the Brainstem: Temporal Relationships
897 MR Imaging of Wallerian Degeneration in the Brainstem: Temporal Relationships Yuichi Inoue 1 Degeneration of the myelin sheath and axon distal to the most proximal site of axonal Yasumasa Matsumura2 interruption secondary to axonal disease has been called wallerian degeneration. On Teruo Fukuda1 MR imaging, wallerian degeneration of the pyramidal tract can be observed as an Yutaka Nemoto1 abnormal signal intensity, showing prolonged T1 and T2 relaxation times that correspond to the corticospinal tract, with or without shrinkage of the ipsilateral cerebral peduncle Nobuyuki Shirahata3 and pons. Review of MR studies in 150 cases of supratentorial cerebrovascular accidents Tosihisa Suzuki4 1 showed abnormal signal alterations in the ipsilateral brainstem in 33 of the cases. Miyuki Shakudo Abnormal intensity in the ipsilateral brainstem was seen as early as 5 weeks after the 1 Satoshi Yawata supratentorial ictus and was fully evident after 10 weeks in all 33 cases. Signal 2 Shigeko Tanaka alterations were strongest at about 3-6 months when compared with alterations seen Kazumasa Takemoto5 at 10 weeks or even 10 months after the ictus. Shrinkage of the ipsilateral brainstem Yasuto Onoyama 1 appeared as early as 8 months and was demonstrated in all cases 13 months after the ictus. MR seems to be the most effective technique for early detection of wallerian degen eration and may provide insight into its pathophysiological and chemical changes. AJNR 11:897-902, September/October 1990 Wallerian degeneration is the process of disintegration that affects an axon and its myelin sheath after its connection with the cell body has been interrupted [1 ). -

The Size and Distribution of Midbrain Dopaminergic Populations Are Permanently Altered by Perinatal Glucocorticoid Exposure in a Sex- Region- and Time-Specific Manner
Neuropsychopharmacology (2007) 32, 1462–1476 & 2007 Nature Publishing Group All rights reserved 0893-133X/07 $30.00 www.neuropsychopharmacology.org The Size and Distribution of Midbrain Dopaminergic Populations are Permanently Altered by Perinatal Glucocorticoid Exposure in a Sex- Region- and Time-Specific Manner Simon McArthur1, Emily McHale1 and Glenda E Gillies*,1 1 Department of Cellular and Molecular Neuroscience, Division of Neuroscience and Mental Health, Imperial College, London, UK Central dopaminergic (DA) systems appear to be particularly vulnerable to disruption by exposure to stressors in early life, but the underlying mechanisms are poorly understood. As endogenous glucocorticoids (GCs) are implicated in other aspects of neurobiological programming, this study aimed to characterize the effects of perinatal GC exposure on the cytoarchitecture of DA populations in the substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA). Dexamethasone was administered non-invasively to rat pups via the mothers’ drinking water during embryonic days 16–19 or postnatal days 1–7, with a total oral intake circa 0.075 or 0.15 mg/ kg/day, respectively; controls received normal drinking water. Analysis of tyrosine hydroxylase-immunoreactive cell counts and regional volumes in adult offspring identified notable sex differences in the shape and volume of the SNc and VTA, as well as the topographical organization and size of the DA populations. Perinatal GC treatments increased the DA population size and altered the shape of the SNc and VTA as well as the organization of the DA neurons by expanding and/or shifting them in a caudal direction. This response was sexually dimorphic and included a feminization or demasculinization of the three-dimensional cytoarchitecture in males, and subtle differences that were dependent on the window of exposure. -

Pilot Longitudinal Study on Post-Stroke Subjects
brain sciences Article Brain Plasticity Mechanisms Underlying Motor Control Reorganization: Pilot Longitudinal Study on Post-Stroke Subjects Marta Gandolla 1,2,* , Lorenzo Niero 1, Franco Molteni 3, Elenora Guanziroli 3 , Nick S. Ward 4,5 and Alessandra Pedrocchi 1,6 1 NearLab@Lecco, Polo Territoriale di Lecco, Politecnico di Milano, Via Gaetano Previati, 1/c, 23900 Lecco, Italy; [email protected] (L.N.); [email protected] (A.P.) 2 Department of Mechanical Engineering, Politecnico di Milano, Via Privata Giuseppe La Masa, 1, 20156 Milano, Italy 3 Villa Beretta Rehabilitation Center, Valduce Hospital, Via N. Sauro, 17, 23845 Costa Masnaga, Italy; [email protected] (F.M.); [email protected] (E.G.) 4 Department of Movement and Clinical Neuroscience, UCL Queen Square Institute of Neurology, London WC1N 3BG, UK; [email protected] 5 The National Hospital for Neurology and Neurosurgery, Queen Square, London WC1N 3BG, UK 6 NearLab, Department of Electronic Information and Bioengineering, Politecnico di Milano, Via Giuseppe Ponzio, 34/5, 20133 Milano, Italy * Correspondence: [email protected] Citation: Gandolla, M.; Niero, L.; Abstract: Functional Electrical Stimulation (FES) has demonstrated to improve walking ability Molteni, F.; Guanziroli, E.; Ward, N.S.; and to induce the carryover effect, long-lasting persisting improvement. Functional magnetic Pedrocchi, A. Brain Plasticity resonance imaging has been used to investigate effective connectivity differences and longitudinal Mechanisms Underlying Motor changes in a group of chronic stroke patients that attended a FES-based rehabilitation program Control Reorganization: Pilot for foot-drop correction, distinguishing between carryover effect responders and non-responders, Longitudinal Study on Post-Stroke and in comparison with a healthy control group. -

Role of P2y Receptors in the Spinal-Trigeminal System in Vivo and in Vitro
UNIVERSITÀ DEGLI STUDI DI MILANO Facoltà di Farmacia Dipartimento di Scienze Farmacologiche Corso di Dottorato di Ricerca in Scienze Farmacotossicologiche, Farmacognostiche e Biotecnologie Farmacologiche (XXIII CICLO) Graduate School in Pharmacological Sciences / Scuola di Dottorato in Scienze farmacologiche TESI DI DOTTORATO DI RICERCA PURINERGIC TRANSMISSION IN MIGRAINE: ROLE OF P2Y RECEPTORS IN THE SPINAL-TRIGEMINAL SYSTEM IN VIVO AND IN VITRO BIO/14 Tesi di dottorato di: GIOVANNI VILLA MATRICOLA: R07517 TUTOR: Chiar.ma Prof.ssa Maria Pia ABBRACCHIO CORRELATORE: Dr.ssa Stefania CERUTI COORDINATORE: Chiar.mo Prof. Guido FRANCESCHINI ANNO ACCADEMICO 2009/2010 Index INDEX 1. INTRODUCTION _______________________________ 1 1.1 PAIN AND NOCICEPTION 2 1.1.1 Molecular basis of nociception 3 1.1.2 The trigeminal nerve and the spinal-trigeminal system 4 1.1.3 Role of non-neuronal cells in pain transmission 9 1.2 MIGRAINE 12 1.2.1 Description of the migraine attack 14 1.2.2 How and where does the migraine attack originate? 15 1.2.3 Familial hemiplegic migraine 21 1.2.4 Current and future pharmacological treatment of migraine 24 1.3 THE PURINERGIC SYSTEM 29 1.3.1 Purinergic signalling 30 1.3.2 P2X receptors 32 1.3.3 P2Y receptors 33 1.3.4 Pathophysiological roles of extracellular nucleotides in the nervous system 36 1.4 PURINES AND PAIN 41 1.4.1 Role of P2X receptors in pain transmission 42 1.4.2 Role of P2Y receptors in pain transmission: sensory ganglia 46 1.4.3 Role of P2Y receptors in pain transmission: CNS 48 2. -
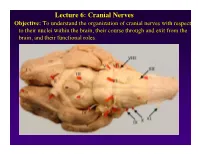
Lecture 6: Cranial Nerves
Lecture 6: Cranial Nerves Objective: To understand the organization of cranial nerves with respect to their nuclei within the brain, their course through and exit from the brain, and their functional roles. Olfactory Eye Muscles 3, 4 &6 Cranial Nerves 1-7 I overview Table, Page 49 II Lecture notes Cranial Nerves and their Functions V Trigeminal VII Facial VIII IX X XII XI Cranial Nerves 8-12 Overview sternocephalic I. Factors Responsible for the Complex Internal Organization of the Brain Stem-> leads to altered location of cranial nerve nuclei in adult brain stem 1. Development of the Fourth Ventricle a. Medulla and Pons develop ventral to the 4th ventricle cerebellum b. Alar plate is displaced lateral to basal plate 4 Medulla Developing Neural Tube 2. Cranial nerve nuclei form discontinuous columns Rostral 12 SE Page 48 Notes 3. Some cranial nerve nuclei migrate from their primitive embryonic positions (e.g., nuclei of V and VII) Facial N. Factors responsible for the complex internal organization of the brainstem: 4) Special senses develop in association with the brain stem. Nuclei of special senses 5) Development of the cerebellum and its connections Cerebellum II. Cranial Nerve Nuclei: Nucleus = column of neuron cell bodies. Efferent nuclei are composed of cell bodies of alpha or gamma motor neurons (SE) or preganglionic parasympathetic neurons (VE). III. Motor Efferent Nuclei (Basal Plate Derivatives): 1. SE (Somatic Efferent) Nuclei: SE neurons form two longitudinally oriented but discontinuous columns of cell bodies in the brain stem. Neurons that comprise these columns are responsible for innervating all of the skeletal musculature of the head. -

The Human Cortical Dental Pain Matrix : Neural Activation Patterns of Tooth Pain Investigated with Fmri
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2011 The human cortical dental pain matrix : neural activation patterns of tooth pain investigated with fMRI Brügger, Michael Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-164035 Dissertation Published Version Originally published at: Brügger, Michael. The human cortical dental pain matrix : neural activation patterns of tooth pain investigated with fMRI. 2011, University of Zurich, Faculty of Arts. The Human Cortical Dental Pain Matrix Neural Activation Patterns of Tooth Pain investigated with fMRI Thesis presented to the Faculty of Arts of the University of Zurich for the degree of Doctor of Philosophy by Michael Brügger of Marbach SG Accepted in the spring semester 2009 on the recommendation of Prof. Dr. rer. nat. Lutz Jäncke and Prof. Dr. med. dent. Sandro Palla Zurich, 2011 …the authors "art‐like" interpretation of a human brain under tooth pain… CONTENTS SUMMARY .................................................................................................................................. 6 ZUSAMMENFASSUNG ................................................................................................................ 7 PREFACE ..................................................................................................................................... 9 1. INTRODUCTION .................................................................................................... -

The High Yield Neurologic Examination Sadly, I Still Have Nothing New to Disclose Since Tuesday John Engstrom, M.D
Disclosures The High Yield Neurologic Examination Sadly, I still have nothing new to disclose since Tuesday John Engstrom, M.D. August 2018 Medical Chart Quotes Medical Chart Quotes “Exam of the genitalia reveals that he is circus sized.” “While in the ER, She was examined, X- rated and sent home.” 1 Overview – The Neurologic Medical Chart Quotes Examination • Mental status-brief review “Both breasts are equal and reactive • Cranial nerves – common/urgent patterns • Motor exam – common patterns to light and accommodation” • Sensory exam – common patterns • What is wrong with my walking? • Demonstrate the 15 minute exam • Other questions/demonstrations? Screening Mental Status Screening for Visual Field Deficits • Orientation-time, place, person • Visual field screen if you suspect a brain problem • Attention-Digit span forward (nl > 6-7) • Allows you to test function of broad areas of brain – Lobes-occipital, temporal, parietal • Language-repetition, naming, comprehen – Optic nerves, chiasm, optic tracts, and thalamus • Memory-Recall of 3 common objects at 5 • Clinical Importance minutes; if misses an answer give a prompt – “An anatomic sedimentation rate of the brain” • Abstractions-Similarities and differences – Detect abnormalities that require brain imaging (e.g.-apple vs. orange; lake vs. river) – Localize the deficit (right vs. left brain) 2 Screening for Visual Field Deficits- Ambulatory, Cooperative Patient • Imagine visual field cut in four equal pieces • Move examiner finger in the center of each quadrant with patient gaze fixed -

A. Diencephalon B. Telencephalon C. Metencephalon D
SAMPLE TEST QUESTIONS Select the one best answer. l. The olfactory nerve is attached to the: A. Diencephalon B. Telencephalon C. Metencephalon D. Mesencephalon E. Myelencephalon 2. Following complete transection of a peripheral nerve, all the following may occur except: A. Chromatoloysis in the cell body. B. Degeneration of peripheral myelin distal to the site of injury. C. Degeneration of the axon distal to the site of injury. D. Immediate restoration of normal function following prompt surgical repair. E. Outgrowth of axonal sprouting from the proximal nerve stump .. 3. A lesion of the left hypoglossal nerve causes: A. Loss of taste sensation on the left side of the tongue. B. Deviation of the tongue to the right side, upon protrusion. C. Total inability to protrude the tongue. D. Deviation of the tongue to the left, upon protrusion. E. Hoarseness. 4. In the brain stem, the principle sensory decussation f0r general sensation ca~ried in the posterior columns of the spinal cord forms the: A. Medial lemniscus. B. Lateral lemniscus. C. Central tegmental fasciculus D. Medial longitudinal fasciculus E. Spinal lemniscus. 5. All are true of the lateral spinothalamic tract except: A. It caries sensations of pain and thermal sense. B. Fibers from sacral origin are located in the ventro-medial portion of the tract. C. Decussation occurs within one or two spinal segments of its origin. D. Fibers of cervical origin are located in the ventro-medial portion of the tract. E. It forms part of the brain stem lemniscal systems. 81 6. A unilateral lesion of the internal capsule involving the genu and the posterior limb, would cause: A.