Ety, 2018, 124, 518–532
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Haida Gwaii Slug,Staala Gwaii
COSEWIC Assessment and Status Report on the Haida Gwaii Slug Staala gwaii in Canada SPECIAL CONCERN 2013 COSEWIC status reports are working documents used in assigning the status of wildlife species suspected of being at risk. This report may be cited as follows: COSEWIC. 2013. COSEWIC assessment and status report on the Haida Gwaii Slug Staala gwaii in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. x + 44 pp. (www.registrelep-sararegistry.gc.ca/default_e.cfm). Production note: COSEWIC would like to acknowledge Kristiina Ovaska and Lennart Sopuck of Biolinx Environmental Research Inc., for writing the status report on Haida Gwaii Slug, Staala gwaii, in Canada, prepared under contract with Environment Canada. This report was overseen and edited by Dwayne Lepitzki, Co-chair of the COSEWIC Molluscs Specialist Subcommittee. For additional copies contact: COSEWIC Secretariat c/o Canadian Wildlife Service Environment Canada Ottawa, ON K1A 0H3 Tel.: 819-953-3215 Fax: 819-994-3684 E-mail: COSEWIC/[email protected] http://www.cosewic.gc.ca Également disponible en français sous le titre Ếvaluation et Rapport de situation du COSEPAC sur la Limace de Haida Gwaii (Staala gwaii) au Canada. Cover illustration/photo: Haida Gwaii Slug — Photo by K. Ovaska. Her Majesty the Queen in Right of Canada, 2013. Catalogue No. CW69-14/673-2013E-PDF ISBN 978-1-100-22432-9 Recycled paper COSEWIC Assessment Summary Assessment Summary – May 2013 Common name Haida Gwaii Slug Scientific name Staala gwaii Status Special Concern Reason for designation This small slug is a relict of unglaciated refugia on Haida Gwaii and on the Brooks Peninsula of northwestern Vancouver Island. -

BRYOLOGICAL INTERACTION-Chapter 4-6
65 CHAPTER 4-6 INVERTEBRATES: MOLLUSKS Figure 1. Slug on a Fissidens species. Photo by Janice Glime. Mollusca – Mollusks Glistening trails of pearly mucous criss-cross mats and also seemed to be a preferred food. Perhaps we need to turfs of green, signalling the passing of snails and slugs on searach at night when the snails and slugs are more active. the low-growing bryophytes (Figure 1). In California, the white desert snail Eremarionta immaculata is more common on lichens and mosses than on other plant detritus and rocks (Wiesenborn 2003). Wiesenborn suggested that the snails might find more food and moisture there. Are these mollusks simply travelling from one place to another across the moist moss surface, or do they have a more dastardly purpose for traversing these miniature forests? Quantitative information on snails and slugs among bryophytes is scarce, and often only mentions that bryophytes are abundant in the habitat (e.g. Nekola 2002), but we might be able to glean some information from a study by Grime and Blythe (1969). In collections totalling 82.4 g of moss, they examined snail populations in a 0.75 m2 plot each morning on 7, 8, 9, & 12 September 1966. The copse snail, Arianta arbustorum (Figure 2), numbered 0, 7, 2, and 6 on those days, respectively, with weights of Figure 2. The copse snail, Arianta arbustorum, in 0.0, 8.5, 2.4, & 7.3 per 100 g dry mass of moss. They were Stockholm, Sweden. Photo by Håkan Svensson through most abundant on the stinging nettle, Urtica dioica, which Wikimedia Commons. -

Comprehensive Conservation Plan Benton Lake National Wildlife
Glossary accessible—Pertaining to physical access to areas breeding habitat—Environment used by migratory and activities for people of different abilities, es- birds or other animals during the breeding sea- pecially those with physical impairments. son. A.D.—Anno Domini, “in the year of the Lord.” canopy—Layer of foliage, generally the uppermost adaptive resource management (ARM)—The rigorous layer, in a vegetative stand; mid-level or under- application of management, research, and moni- story vegetation in multilayered stands. Canopy toring to gain information and experience neces- closure (also canopy cover) is an estimate of the sary to assess and change management activities. amount of overhead vegetative cover. It is a process that uses feedback from research, CCP—See comprehensive conservation plan. monitoring, and evaluation of management ac- CFR—See Code of Federal Regulations. tions to support or change objectives and strate- CO2—Carbon dioxide. gies at all planning levels. It is also a process in Code of Federal Regulations (CFR)—Codification of which the Service carries out policy decisions the general and permanent rules published in the within a framework of scientifically driven ex- Federal Register by the Executive departments periments to test predictions and assumptions and agencies of the Federal Government. Each inherent in management plans. Analysis of re- volume of the CFR is updated once each calendar sults helps managers decide whether current year. management should continue as is or whether it compact—Montana House bill 717–Bill to Ratify should be modified to achieve desired conditions. Water Rights Compact. alternative—Reasonable way to solve an identi- compatibility determination—See compatible use. -

2010 Animal Species of Concern
MONTANA NATURAL HERITAGE PROGRAM Animal Species of Concern Species List Last Updated 08/05/2010 219 Species of Concern 86 Potential Species of Concern All Records (no filtering) A program of the University of Montana and Natural Resource Information Systems, Montana State Library Introduction The Montana Natural Heritage Program (MTNHP) serves as the state's information source for animals, plants, and plant communities with a focus on species and communities that are rare, threatened, and/or have declining trends and as a result are at risk or potentially at risk of extirpation in Montana. This report on Montana Animal Species of Concern is produced jointly by the Montana Natural Heritage Program (MTNHP) and Montana Department of Fish, Wildlife, and Parks (MFWP). Montana Animal Species of Concern are native Montana animals that are considered to be "at risk" due to declining population trends, threats to their habitats, and/or restricted distribution. Also included in this report are Potential Animal Species of Concern -- animals for which current, often limited, information suggests potential vulnerability or for which additional data are needed before an accurate status assessment can be made. Over the last 200 years, 5 species with historic breeding ranges in Montana have been extirpated from the state; Woodland Caribou (Rangifer tarandus), Greater Prairie-Chicken (Tympanuchus cupido), Passenger Pigeon (Ectopistes migratorius), Pilose Crayfish (Pacifastacus gambelii), and Rocky Mountain Locust (Melanoplus spretus). Designation as a Montana Animal Species of Concern or Potential Animal Species of Concern is not a statutory or regulatory classification. Instead, these designations provide a basis for resource managers and decision-makers to make proactive decisions regarding species conservation and data collection priorities in order to avoid additional extirpations. -

Characterization of Arm Autotomy in the Octopus, Abdopus Aculeatus (D’Orbigny, 1834)
Characterization of Arm Autotomy in the Octopus, Abdopus aculeatus (d’Orbigny, 1834) By Jean Sagman Alupay A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Integrative Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Roy L. Caldwell, Chair Professor David Lindberg Professor Damian Elias Fall 2013 ABSTRACT Characterization of Arm Autotomy in the Octopus, Abdopus aculeatus (d’Orbigny, 1834) By Jean Sagman Alupay Doctor of Philosophy in Integrative Biology University of California, Berkeley Professor Roy L. Caldwell, Chair Autotomy is the shedding of a body part as a means of secondary defense against a predator that has already made contact with the organism. This defense mechanism has been widely studied in a few model taxa, specifically lizards, a few groups of arthropods, and some echinoderms. All of these model organisms have a hard endo- or exo-skeleton surrounding the autotomized body part. There are several animals that are capable of autotomizing a limb but do not exhibit the same biological trends that these model organisms have in common. As a result, the mechanisms that underlie autotomy in the hard-bodied animals may not apply for soft bodied organisms. A behavioral ecology approach was used to study arm autotomy in the octopus, Abdopus aculeatus. Investigations concentrated on understanding the mechanistic underpinnings and adaptive value of autotomy in this soft-bodied animal. A. aculeatus was observed in the field on Mactan Island, Philippines in the dry and wet seasons, and compared with populations previously studied in Indonesia. -

Prophysaon Coeruleum Conservation Assessment
CONSERVATION ASSESSMENT FOR Prophysaon coeruleum, Blue-Gray Taildropper Originally issued as Management Recommendations September 1999 by Thomas E. Burke with contributions by Nancy Duncan and Paul Jeske Reconfigured October 2005 Nancy Duncan USDA Forest Service Region 6 and USDI Bureau of Land Management, Oregon and Washington TABLE OF CONTENTS EXECUTIVE SUMMARY ......................................................................................................... 1 I. NATURAL HISTORY ................................................................................................... 3 A. Taxonomic/Nomenclatural History ...................................................................... 3 B. Species Description ............................................................................................... 3 1. Morphology ............................................................................................... 3 2. Reproductive Biology ................................................................................ 5 3. Ecology ...................................................................................................... 5 C. Range, Known Sites................................................................................................ 6 D. Habitat Characteristics and Species Abundance..................................................... 6 1. Habitat Characteristics ............................................................................... 6 2. Species Abundance ................................................................................... -
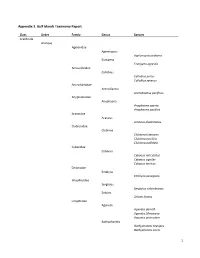
1 Appendix 3. Gulf Islands Taxonomy Report
Appendix 3. Gulf Islands Taxonomy Report Class Order Family Genus Species Arachnida Araneae Agelenidae Agelenopsis Agelenopsis utahana Eratigena Eratigena agrestis Amaurobiidae Callobius Callobius pictus Callobius severus Antrodiaetidae Antrodiaetus Antrodiaetus pacificus Anyphaenidae Anyphaena Anyphaena aperta Anyphaena pacifica Araneidae Araneus Araneus diadematus Clubionidae Clubiona Clubiona lutescens Clubiona pacifica Clubiona pallidula Cybaeidae Cybaeus Cybaeus reticulatus Cybaeus signifer Cybaeus tetricus Dictynidae Emblyna Emblyna peragrata Gnaphosidae Sergiolus Sergiolus columbianus Zelotes Zelotes fratris Linyphiidae Agyneta Agyneta darrelli Agyneta fillmorana Agyneta protrudens Bathyphantes Bathyphantes brevipes Bathyphantes keeni 1 Centromerita Centromerita bicolor Ceratinops Ceratinops latus Entelecara Entelecara acuminata Erigone Erigone aletris Erigone arctica Erigone cristatopalpus Frederickus Frederickus coylei Grammonota Grammonota kincaidi Linyphantes Linyphantes nehalem Linyphantes nigrescens Linyphantes pacificus Linyphantes pualla Linyphantes victoria Mermessus Mermessus trilobatus Microlinyphia Microlinyphia dana Neriene Neriene digna Neriene litigiosa Oedothorax Oedothorax alascensis Pityohyphantes Pityohyphantes alticeps Pocadicnemis Pocadicnemis pumila Poeciloneta Poeciloneta fructuosa Saaristoa Saaristoa sammamish Scotinotylus Scotinotylus sp. 5GAB Semljicola Semljicola sp. 1GAB Sisicottus Spirembolus Spirembolus abnormis Spirembolus mundus Tachygyna Tachygyna ursina Tachygyna vancouverana Tapinocyba Tapinocyba -

Abstract Volume
ABSTRACT VOLUME August 11-16, 2019 1 2 Table of Contents Pages Acknowledgements……………………………………………………………………………………………...1 Abstracts Symposia and Contributed talks……………………….……………………………………………3-225 Poster Presentations…………………………………………………………………………………226-291 3 Venom Evolution of West African Cone Snails (Gastropoda: Conidae) Samuel Abalde*1, Manuel J. Tenorio2, Carlos M. L. Afonso3, and Rafael Zardoya1 1Museo Nacional de Ciencias Naturales (MNCN-CSIC), Departamento de Biodiversidad y Biologia Evolutiva 2Universidad de Cadiz, Departamento CMIM y Química Inorgánica – Instituto de Biomoléculas (INBIO) 3Universidade do Algarve, Centre of Marine Sciences (CCMAR) Cone snails form one of the most diverse families of marine animals, including more than 900 species classified into almost ninety different (sub)genera. Conids are well known for being active predators on worms, fishes, and even other snails. Cones are venomous gastropods, meaning that they use a sophisticated cocktail of hundreds of toxins, named conotoxins, to subdue their prey. Although this venom has been studied for decades, most of the effort has been focused on Indo-Pacific species. Thus far, Atlantic species have received little attention despite recent radiations have led to a hotspot of diversity in West Africa, with high levels of endemic species. In fact, the Atlantic Chelyconus ermineus is thought to represent an adaptation to piscivory independent from the Indo-Pacific species and is, therefore, key to understanding the basis of this diet specialization. We studied the transcriptomes of the venom gland of three individuals of C. ermineus. The venom repertoire of this species included more than 300 conotoxin precursors, which could be ascribed to 33 known and 22 new (unassigned) protein superfamilies, respectively. Most abundant superfamilies were T, W, O1, M, O2, and Z, accounting for 57% of all detected diversity. -

Integrating Life History Traits Into Predictive Phylogeography
Received: 20 August 2018 | Revised: 4 January 2019 | Accepted: 16 January 2019 DOI: 10.1111/mec.15029 ORIGINAL ARTICLE Integrating life history traits into predictive phylogeography Jack Sullivan1,2* | Megan L. Smith3* | Anahí Espíndola1,4 | Megan Ruffley1,2 | Andrew Rankin1,2 | David Tank1,2 | Bryan Carstens3 1Department of Biological Sciences, University of Idaho, Moscow, Abstract Idaho Predictive phylogeography seeks to aggregate genetic, environmental and taxonomic 2 Institute for Bioinformatics and data from multiple species in order to make predictions about unsampled taxa using Evolutionary Studies, University of Idaho, Moscow, Idaho machine‐learning techniques such as Random Forests. To date, organismal trait data 3Department of Ecology, Evolution and have infrequently been incorporated into predictive frameworks due to difficulties Organismal Biology, The Ohio State University, Columbus, Ohio inherent to the scoring of trait data across a taxonomically broad set of taxa. We re‐ 4Department of Entomology, University of fine predictive frameworks from two North American systems, the inland temperate Maryland, College Park, Maryland rainforests of the Pacific Northwest and the Southwestern Arid Lands (SWAL), by Correspondence incorporating a number of organismal trait variables. Our results indicate that incor‐ Jack Sullivan, Department of Biological porating life history traits as predictor variables improves the performance of the Sciences, University of Idaho, Moscow, ID. Email: [email protected] supervised machine‐learning approach to predictive phylogeography, especially for Funding information the SWAL system, in which predictions made from only taxonomic and climate vari‐ National Science Foundation, Grant/Award ables meets only moderate success. In particular, traits related to reproduction (e.g., Numbers: DEB 14575199, DEB 1457726, DG‐1343012; NSF GRFP; Ohio State reproductive mode; clutch size) and trophic level appear to be particularly informa‐ University; Institute for Bioinformatics and tive to the predictive framework. -

Determining the Occurrence and Range of Deroceras Hesperium On
FY2012 ISSSP Report on Determining the Occurrence and Range of Deroceras hesperium* on the Willamette National Forest using Summer Surveys of Unique Habitats and Comparison of Summer Surveys versus Spring/Fall Mollusk Protocol Surveys for Detection of this Species Authored by Tiffany Young and Joe Doerr (Willamette National Forest). Dated 08/21/13. [*Note on Taxonomy: During the course of our study, a genetic investigation concluded that the Deroceras hesperium (the evening field slug) should be consider a junior synonym of Deroceras laeve (the meadow slug) (Roth et al. 2013). Our report, based on a study that began when two species were recognized, continues to use the original convention of referring to Deroceras with tripartite soles as D. hesperium and those with uniformly colored soles as D. laeve. We recognize the difficulty in making this distinction and our usage of the 2 species is not intended to imply that we consider them separate species. Moreover, in our study, both types used similar habitat and we have assumed that all detections help define the habitat preferences of both types.] Introduction: Prior to Roth et al. (2013,), Deroceras hesperium (DEHE) was considered a rare species in Region 6 with few documented locations at scattered sites in western Oregon. In 2008 it was listed as a “suspected” sensitive species on the Willamette NF because it had been found at nearby locations on Salem BLM. However, it has never been detected on the Willamette Forest itself, despite more than a decade of project mollusk surveys. Deroceras hesperium is also managed as a Category B sensitive species in Region 6 that requires equivalent effort surveys for habitat disturbing actions. -

Montana's State Wildlife Action Plan 2015
MONTANA’S STATE WILDLIFE ACTION PLAN MONTANA FISH, WILDLIFE & PARKS 2015 The mission of Montana Fish, Wildlife & Parks (FWP) is to provide for the stewardship of the fish, wildlife, parks, and recreational resources of Montana, while contributing to the quality of life for present and future generations. To carry out its mission, FWP strives to provide and support fiscally responsible programs that conserve, enhance, and protect Montana’s 1) aquatic ecotypes, habitats, and species; 2) terrestrial ecotypes, habitats, and species; and 3) important cultural and recreational resources. This document should be cited as Montana’s State Wildlife Action Plan. 2015. Montana Fish, Wildlife & Parks, 1420 East Sixth Avenue, Helena, MT 59620. 441 pp. EXECUTIVE SUMMARY Montana’s first State Wildlife Action Plan (SWAP), the Comprehensive Fish and Wildlife Conservation Strategy (CFWCS), was approved by the U.S. Fish and Wildlife Service in 2006. Since then, many conservation partners have used the plan to support their conservation work and to seek additional funding to continue their work. For Montana Fish, Wildlife & Parks (FWP), State Wildlife Grant (SWG) dollars have helped implement the strategy by supporting conservation efforts for many different species and habitats. This revision details implemented actions since 2006 (Appendix C). This SWAP identifies community types, Focal Areas, and species in Montana with significant issues that warrant conservation attention. The plan is not meant to be an FWP plan, but a plan to guide conservation throughout Montana. One hundred and twenty-eight Species of Greatest Conservation Need (SGCN) are identified in this revision. Forty-seven of these are identified as being in most critical conservation need. -

Terrestrial Mollusk Surveys on Region 1 USFS Lands
Terrestrial Mollusk Surveys on Region 1 USFS Lands Paul Hendricks, Bryce Maxell, Susan Lenard, Coburn Currier, and Ryan Kilacky Montana Natural Heritage Program http://nhp.nris.state.mt.us Globally Rare Land Snails Present on R1 Forests • Selway Forestsnail (Allogona lombardii) (ID) G1 • Dry Land Forestsnail (Allogona ptychophora solida) (ID)? G5T2T3 • Nimapuna Tigersnail (Anguispira nimapuna) (ID) G1 • Chrome Ambershell (Catinella rehderi) (MT, ID?) G1G2Q* • Salmon Oregonian (Cryptomastix harfordiana) (ID)? G3G4 • Mission Creek Oregonian (Cryptomastix magnidentata) (ID)? G1 • Oregonian (Cryptomastix mullani blandi) (ID)? G4T1 • River of No Return Oregonian (Cryptomastix mullani clappi) (ID) G4T1 • Kingston Oregonian (Cryptomastix sanburni) (ID)? G1 SUMMARY • Lake Disc (Discus brunsoni) (MT)? G1 • Marbled Disc (Discus marmorensis) (ID) G1G3 • 31 Species G1-G3 so USFS SOC • Striate Disc (Discus shimekii) (MT, ID?) G5 • 2 Species G5, but S1-S3 so USFS SOI • Salmon Coil (Helicodiscus salmonaceus) (ID) G1G2 • Alpine Mountainsnail (Oreohelix alpina) (MT) G1 • Bitterroot Mountainsnail (Oreohelix amariradix) (MT) G1G2 • Keeled Mountainsnail (Oreohelix carinifera) (MT) G1 • Carinate Mountainsnail (Oreohelix elrodi) (MT) G1 • Seven Devils Mountainsnail (Oreohelix hammeri) (ID) G1 • A Land Snail (Hells Canyon) (Oreohelix idahoensis baileyi) (ID) G1G2T1 • Costate Mountainsnail (Oreohelix idahoensis idahoensis) (ID)? G1G2T1T2 • Deep Slide Mountainsnail (Oreohelix intersum) (ID)? G1 • Boulder Pile Mountainsnail (Oreohelix jugalis) (ID)? G1 • Berry’s