The Devulcanization of Unfilled and Carbon Black Filled
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Little Book About BIG Chemistry the Story of Man-Made Polymers
SPRINGER BRIEFS IN MATERIALS Jim Massy A Little Book about BIG Chemistry The Story of Man-Made Polymers www.ebook3000.com SpringerBriefs in Materials The SpringerBriefs Series in Materials presents highly relevant, concise mono- graphs on a wide range of topics covering fundamental advances and new applications in the field. Areas of interest include topical information on innovative, structural and functional materials and composites as well as fundamental principles, physical properties, materials theory and design. SpringerBriefs present succinct summaries of cutting-edge research and practical applications across a wide spectrum of fields. Featuring compact volumes of 50 to 125 pages, the series covers a range of content from professional to academic. Typical topics might include: • A timely report of state-of-the-art analytical techniques • A bridge between new research results, as published in journal articles, and a contextual literature review • A snapshot of a hot or emerging topic • An in-depth case study or clinical example • A presentation of core concepts that students must understand in order to make independent contributions Briefs are characterized by fast, global electronic dissemination, standard publishing contracts, standardized manuscript preparation and formatting guide- lines, and expedited production schedules. More information about this series at http://www.springer.com/series/10111 www.ebook3000.com Jim Massy A Little Book about BIG Chemistry The Story of Man-Made Polymers 123 Jim Massy Norwich, Norfolk UK ISSN 2192-1091 ISSN 2192-1105 (electronic) SpringerBriefs in Materials ISBN 978-3-319-54830-2 ISBN 978-3-319-54831-9 (eBook) DOI 10.1007/978-3-319-54831-9 Library of Congress Control Number: 2017934463 © The Author(s) 2017 This work is subject to copyright. -

Vulcanization & Accelerators
Vulcanization & Accelerators Vulcanization is a cross linking process in which individual molecules of rubber (polymer) are converted into a three dimensional network of interconnected (polymer) chains through chemical cross links(of sulfur). The vulcanization process was discovered in 1839 and the individuals responsible for this discovery were Charles Goodyear in USA and Thomas Hancock in England. Both discovered the use of Sulfur and White Lead as a vulcanization system for Natural Rubber. This discovery was a major technological breakthrough for the advancement of the world economy. Vulcanization of rubbers by sulfur alone is an extremely slow and inefficient process. The chemical reaction between sulfur and the Rubber Hydrocarbon occurs mainly at the C = C (double bonds) and each crosslink requires 40 to 55 sulphur atoms (in the absence of accelerator). The process takes around 6 hours at 140°C for completion, which is uneconomical by any production standards. The vulcanizates thus produced are extremely prone to oxidative degradation and do not possess adequate mechanical properties for practical rubber applications. These limitations were overcome through inventions of accelerators which subsequently became a part of rubber compounding formulations as well as subjects of further R&D. Following is the summary of events which led to the progress of ‘Accelerated Sulfur Vulcanization'. Event Year Progress - Discovery of Sulfur Vulcanization: Charles Goodyear. 1839 Vulcanizing Agent - Use of ammonia & aliphatic ammonium derivatives: Rowley. 1881 Acceleration need - Use of aniline as accelerator in USA & Germany: Oenslager. 1906 Accelerated Cure - Use of Piperidine accelerator- Germany. 1911 New Molecules - Use of aldehyde-amine & HMT as accelerators in USA & UK 1914-15 Amine Accelerators - Use of Zn-Alkyl Xanthates accelerators in Russia. -

Zinc Complexes with 1,3-Diketones As Activators for Sulfur Vulcanization of Styrene-Butadiene Elastomer Filled with Carbon Black
materials Article Zinc Complexes with 1,3-Diketones as Activators for Sulfur Vulcanization of Styrene-Butadiene Elastomer Filled with Carbon Black Magdalena Maciejewska * , Anna Sowi ´nska* and Agata Grocholewicz Department of Chemistry, Institute of Polymer and Dye Technology, Lodz University of Technology, Stefanowskiego Street 12/16, 90-924 Lodz, Poland; [email protected] * Correspondence: [email protected] (M.M.); [email protected] (A.S.) Abstract: Zinc oxide nanoparticles (N-ZnO) and zinc complexes with 1,3-diketones of different structures were applied instead of microsized zinc oxide (M-ZnO) to activate the sulfur vulcanization of styrene-butadiene rubber (SBR). The influence of vulcanization activators on the cure characteristics of rubber compounds, as well as crosslink density and functional properties of SBR vulcanizates, such as tensile properties, hardness, damping behavior, thermal stability and resistance to thermo- oxidative aging was explored. Applying N-ZnO allowed to reduce the content of zinc by 40% compared to M-ZnO without detrimental influence on the cure characteristic and performance of SBR composites. The activity of zinc complexes in vulcanization seems to strongly depend on their structure, i.e., availability of zinc to react with curatives. The lower the steric hindrance of the substituents and thus the better the availability of zinc ions, the greater was the activity of the zinc complex and consequently the higher the crosslink density of the vulcanizates. Zinc complexes had no Citation: Maciejewska, M.; detrimental effect on the time and temperature of SBR vulcanization. Despite lower crosslink density, Sowi´nska,A.; Grocholewicz, A. Zinc most vulcanizates with zinc complexes demonstrated similar or improved functional properties in Complexes with 1,3-Diketones as Activators for Sulfur Vulcanization of comparison with SBR containing M-ZnO. -

©2020 Yu Sun All Rights Reserved
©2020 YU SUN ALL RIGHTS RESERVED VULCANIZATION AND DEVULCANIZATION OF RUBBER A Dissertation Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirement for the Degree Doctor of Philosophy YU SUN August, 2020 VULCANIZATION AND DEVULCANIZATION OF RUBBER YU SUN Dissertation Approved: Accepted: _____________________________ _____________________________ Advisor Department Chair Dr. Li Jia Dr. Tianbo Liu _____________________________ _____________________________ Committee Chair Interim Dean of the College Dr. Toshikazu Miyoshi Dr. Ali Dhinojwala _____________________________ _____________________________ Committee Member Acting Dean of the Graduate School Dr. Gary R. Hamed Dr. Marnie Saunders _____________________________ _____________________________ Committee Member Date Dr. Junpeng Wang _____________________________ Committee Member Dr. Kevin Cavicchi ii ABSTRACT The research in this dissertation includes two parts: vulcanization of isobutylene- isoprene rubber (IIR) containing geminal vinylidene-acrylate groups and surface devulcanization of ground rubber particles (GRPs) for rubber recycling. In Chapter 2, EIIR was synthesized by grafting ethyl propiolate to IIR via EtAlCl2- catalyzed Alder-ene reaction. Methods for crosslinking EIIR using dicumyl peroxide (DCP) with m-Phenylene-N,N’-bismaleimide (BMI) as a coagent were investigated. The strength, extensibility, and overall toughness of the peroxide-cured EIIR significantly exceed those of previously reported peroxide-cured IIR derivatives. A wide range of stiffness can be realized without sacrificing the strength. The crosslinking density achieved using such curatives is indexed as a function of DCP/BMI ratio, and the curing efficiency of peroxide as a function of DCP/BMI ratio and DCP loading. The role of the geminal vinylidene-acrylate moiety in curing was studied by model reactions, which suggest that the geminal vinylidene-acrylate moiety undergoes both radical addition and hydrogen abstraction. -

Reactive Sintering of Ground Tire Rubber (GTR) Modified by a Trans-Polyoctenamer Rubber and Curing Additives
polymers Article Reactive Sintering of Ground Tire Rubber (GTR) Modified by a Trans-Polyoctenamer Rubber and Curing Additives Łukasz Zedler 1,* , Daria Kowalkowska-Zedler 2 , Xavier Colom 3 , Javier Cañavate 3 , Mohammad Reza Saeb 4 and Krzysztof Formela 1,* 1 Department of Polymer Technology, Faculty of Chemistry, Gda´nskUniversity of Technology, Gabriela, Narutowicza 11/12, 80–233 Gda´nsk,Poland 2 Department of Inorganic Chemistry, Faculty of Chemistry, Gda´nskUniversity of Technology, Gabriela, Narutowicza 11/12, 80–233 Gda´nsk,Poland; [email protected] 3 Department of Chemical Engineering, Universitat Politècnica de Catalunya Barcelona Tech, Carrer de Colom, 1, 08222 Terrassa, Barcelona, Spain; [email protected] (X.C.); [email protected] (J.C.) 4 Center of Excellence in Electrochemistry, School of Chemistry, College of Science, University of Tehran, Tehran 11155-4563, Iran; [email protected] * Correspondence: [email protected] (Ł.Z.); [email protected] (K.F.) Received: 17 November 2020; Accepted: 14 December 2020; Published: 17 December 2020 Abstract: The proposed method of ground tire rubber (GTR) utilization involves the application of trans-polyoctenamer rubber (TOR), a commercially available waste rubber modifier. The idea was to investigate the influence of various curing additives (sulfur, N-cyclohexyl-2-benzothiazole sulfenamide (CBS), dibenzothiazole disulfide (MBTS) and di-(2-ethyl)hexylphosphorylpolysulfide (SDT)) on curing characteristics, physico-mechanical, thermal, acoustic properties as well as the morphology of modified GTR, in order to evaluate the possibility of reclaiming GTR and the co-cross-linking between applied components. The results showed that the presence of the modifier without the addition of curing additives hinders the physico-mechanical properties of revulcanized GTR. -
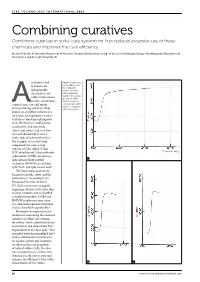
Combining Curatives Combining Curatives in Sulfur Cure Systems for Tires Reduces Excessive Use of These Chemicals and Improves the Cure Efficiency
Combining curatives Combining curatives in sulfur cure systems for tires reduces excessive use of these chemicals and improves the cure efficiency by Saad H Sheikh, Ali Ansarifar (Department of Materials), Jonathan Dushyanthan, George W Weaver and Kahagala Gamage Upul Wijayantha (Department of Chemistry), Loughborough University, UK ccelerators and Figure 1: Cure traces (torque [dNm] versus activators are time [min]) of the indispensable rubber compounds chemicals in the with an increasing loading of the powder. sulfur vulcanization a) compound with of tires. Accelerators 0.63phr powder; b) control onset, rate and extent compound with 2.5phr A powder; c) compound of cross-linking and hence final with 5.6phr powder properties of rubber vulcanizates. Activators are ingredients used to activate accelerators and improve their effectiveness.1 Sulfenamide accelerators, and zinc oxide (ZnO) and stearic acid activators are used extensively to cure a wide range of industrial articles.2 For example, in tire belt skim compound, the cure system consists of 5phr sulfur; 0.7phr N,N’-dicyclohexyl-2-benzothiazole sulfenamide (DCBS) accelerator; 2phr hexamethoxy methyl 1a melamine (HMMM) accelerator; 7phr ZnO; and 1phr stearic acid.3 ZnO and stearic acid can be harmful to health, safety and the environment. According to the European Directive 2004/73/ EC, ZnO is very toxic to aquatic organisms. Stearic acid causes skin and eye irritation and is classified as highly flammable.4 DCBS and HMMM accelerators may cause eye, skin and respiratory irritation and are harmful to aquatic life.5,6 Previously we reported a new method for measuring the chemical curatives in sulfur cure systems for rubber, which eliminated stearic acid entirely and reduced the ZnO requirement to less than 1phr.7 This extended work functionalized ZnO with a sulfenamide accelerator in an organic solvent and examined its effect on the cure properties of natural rubber (NR). -

Vulcanization Kinetics of Graphene/Styrene Butadiene Rubber Nanocomposites*
Chinese Journal of Polymer Science Vol. 32, No. 5, (2014), 658−666 Chinese Journal of Polymer Science © Chinese Chemical Society Institute of Chemistry, CAS Springer-Verlag Berlin Heidelberg 2014 Vulcanization Kinetics of Graphene/Styrene Butadiene Rubber * Nanocomposites Mao-zhu Tang, Wang Xing, Jin-rong Wu**, Guang-su Huang**, Hui Li and Si-duo Wu State Key Laboratory of Polymer Material Engineering, College of Polymer Science and Engineering, Sichuan University, Chengdu 610065, China Abstract This paper presents the influence of graphene on the vulcanization kinetics of styrene butadiene rubber (SBR) with dicumyl peroxide. A curemeter and a differential scanning calorimeter were used to investigate the cure kinetics, from which the kinetic parameters and apparent activation energy were obtained. It turns out that with increasing graphene loading, the induction period of the vulcanization process of SBR is remarkably reduced at low graphene loading and then levels off; on the other hand, the optimum cure time shows a monotonous decrease. As a result, the vulcanization rate is suppressed at first and then accelerated, and the corresponding activation energy increases slightly at first and then decreases. Upon adding graphene, the crosslinking density of the nanocomposites increases, because graphene takes part in the vulcanization process. Keywords: Graphene; Styrene butadiene rubber; Vulcanization kinetics. INTRODUCTION As we all know, the mechanical properties of rubbers strongly rely on the vulcanization state. Therefore, the vulcanization reactions and kinetics of unfilled rubbers had been intensively investigated and well established decades of years ago[1]. However, with the rapid development in nanocomposites, some recent studies have shown that the vulcanization kinetics of rubbers could be changed significantly by the incorporation of nanofillers. -

Rubber Chemistry
RUBBER CHEMISTRY MATADOR RUBBER s.r.o. Summary Rubbers - elastomers - are polymeric materials characterised by their ability of reversible deformation due to external deforming forces. Their deformation rate depends on the structure and molar mass of the deformed rubber and on external conditions of the deformation. This characteristics, referred to as elastic and/or hyper elastic deformation, is entropic in nature and results from the ability of rubber macromolecules to form a more organised state under influence of deforming forces without deformation of chemical bonds between atoms of the polymer chain or without deformation of their valence angles. Ideally, the macromolecules can restore their initial position once the deforming forces are removed. Rubbers usually have long and regular macromolecule chains without bulk substitutes with spatially oriented structural units. This is the reason why their segments are movable and able to rotate around simple chemical bonds even at low temperatures, as it can be seen in their low vitrification temperature Tg. They are tough and similar to plastomers below the vitrification temperature or crystallisation temperature (if rubber can be crystallised). When heated, rubbers change their elastic and/or hyper elastic state to a visco-elastic state; and they become plastic and flow above the softening temperature (Tm). It is advantageous if rubbers crystallise at normal temperature only when subjected to voltage and their Tg is significantly lower that the temperature they are used at. Natural rubber comes from a plant. In industrial applications, it is obtained primarily from Hevea Brasiliensis tree grown in orchards in South-East Asia, Western Africa and northern parts of Southern America. -

Chemical Modification of Bacterial Elastomers: 2. Sulfur Vulcanization
Chemical modification of bacterial elastomers: 2. Sulfur vulcanization K. D. Gagnon*, R. W. Lenz# and R. J. Farris Polymer Science and Engineering Department, University of Massachusetts, Amherst, MA 01003, USA and R. C. Fuller Biochemistry and Molecular Biology Department, University of Massachusetts, Amherst, MA 01003, USA (Received 10 December 1993) Enhancement in the elastic response of a bacterial thermoplastic elastomer, poly(fl-hydroxyoctanoic-co- undecylenic acid), containing varying amounts of unsaturation was attempted through sulfur vulcanization. Sol-gel analysis verified that crosslinking had occurred and that crosslinking varied with vulcanizing agent type and concentration. Differential scanning calorimetry showed that crosslinking could eliminate all crystallinity. The elastic response was near ideal with less than a 5% tensile set after 200% elongation. In general, sulfur-vulcanized materials exhibited a decrease in tensile modulus, tensile strength and tear resistance which did not appear to vary with crosslink density. Unlike the case of peroxide crosslinking, material integrity was left intact and mechanical property results are reported. The network structure was elucidated through the determination of the molecular weight between crosslinks. (Keywords: sulfur vulcanization; crosslinking; microbial polyester) INTRODUCTION have been caused in part by chain scission, a common problem with free radical chemistry. Many of the microbial polyesters produced by Pseudo- The use of sulfur crosslinking or vulcanization was monas oleovorans have the properties of thermoplastic then attempted and is the topic of this report. An elastomers (TPEs). 1 One such elastomeric copolymer, olefin-containing polymer is essential for sulfur cross- poly(#-hydroxyoctanoate) (PHO), has been studied ex- linking. Fortunately, Pseudomonas oleovorans can pro- tensively in our laboratory. -

Vulcanization Kinetics and Mechanical Properties of Ethylene Propylene Diene Monomer Thermal Insulation
Available online at BCREC Website: http://bcrec.undip.ac.id Bulletin of Chemical Reaction Engineering & Catalysis, 10 (2), 2015, 104-110 Research Article Vulcanization Kinetics and Mechanical Properties of Ethylene Propylene Diene Monomer Thermal Insulation Mohamad I. Fathurrohman1*, Dadi R. Maspanger1, S. Sutrisno2 1 Indonesian Rubber Research Institute, Jl. Salak No. 1, Bogor16151, Indonesia 2 Rocket Technology Center, National Institute of Aeronautics and Space, Jl. Raya LAPAN No. 2, Mekarsari, Rumpin, Bogor 16350, Indonesia Received: 8th April 2014; Revised: 7th January 2015; Accepted: 16th January 2015 Abstract The vulcanization kinetics of Ethylene-propylene diene monomer (EPDM) rubber thermal insulation was studied by using rheometer under isothermal condition at different temperatures. The rheometry analysis was used to determine the cure kinetic parameters and predicting the optimum curing time of EPDM thermal insulation. The experimental results revealed that the curing curves of EPDM thermal insulation were marching and the optimum curing time decreased with the increasing of temperature. The kinetic parameters were determined from the autocatalytic model that showed a close fitting with the experimental results. It also indicated a suitability of autocatalytic model in characterizing the cure kinetics. The optimum curing time was predicted from autocatalytic model and the kinetic pa- rameters were obtained by using the relationship between degree of conversion, cure temperature, and cure time. The predictions of cure time provided information on the actual curing characteristic of EPDM thermal insulation. The mechanical properties of EPDM thermal insulation with different vul- canization temperatures showed the same hardness, tensile strength and modulus at 300%, except at temperature 70 oC, while the elongation at breaking point decreased with the increasing temperature of vul- canization. -

Post-Consumer Tires Back Into New Tires, De-Vulcanization and Re-Utilization of Passenger Car Tires
POST-CONSUMER TIRES BACK INTO NEW TIRES DE-VULCANIZATION AND RE-UTILIZATION OF PASSENGER CAR TIRES The research described in this thesis was financially supported by RecyBem B.V., the Netherlands. Graduation committee Chairman: Prof. Dr. F. Eising University of Twente, CTW Secretary: Prof. Dr. F. Eising Promoter: Prof. Dr. Ir. J.W.M. Noordermeer University of Twente, CTW Asst. Promoter: Dr. Ir. W.K. Dierkes University of Twente, CTW Members: Prof. Dr. Ir. M.M.C.G. Warmoeskerken University of Twente, CTW Prof. Dr. Ir. S.R.A. Kersten University of Twente, TNW Prof. Dr. F. Picchioni University of Groningen Prof. Dr. N. Vennemann University of Applied Sciences Osnabrück, Germany Referee: Dr. Ir. L.-Ph.A.E.M. Reuvekamp Apollo Tyres Global R&D, BV Post-consumer tires back into new tires, de-vulcanization and re-utilization of passenger car tires By Sitisaiyidah Saiwari Ph.D. Thesis, University of Twente, Enschede, the Netherlands, 2013. With references ─ With summary in English, Dutch and Thai Copy right © Sitisaiyidah Saiwari, 2013. All rights reserved. Cover design by Panut Kanong & Nara Samran, Yala, Thailand Printed at Wöhrmann Print Service, Postbus 92, 7200 AB Zutphen, the Netherlands. ISBN : 978-90-365-3541-0 DOI: 10.3990/1.9789036535410 URL: http://dx.doi.org/10.3990/1.9789036535410 POST-CONSUMER TIRES BACK INTO NEW TIRES DE-VULCANIZATION AND RE-UTILIZATION OF PASSENGER CAR TIRES DISSERTATION to obtain the degree of doctor at the University of Twente, on the authority of the rector magnificus, Prof. Dr. H. Brinksma, on account of the decision of the graduation committee, to be publicly defended on Thursday, May 23rd, 2013 at 16:45 by Sitisaiyidah Saiwari born on April 1st, 1979 in Mecca, Saudi-Arabia This dissertation has been approved by : Promoter : Prof. -

The Influence of Curing Systems on the Cure Characteristics And
materials Article The Influence of Curing Systems on the Cure Characteristics and Physical Properties of Styrene–Butadiene Elastomer Magdalena Maciejewska * and Monika Siwek Institute of Polymer and Dye Technology, Lodz University of Technology, Stefanowskiego Street 12/16, 90-924 Lodz, Poland; [email protected] * Correspondence: [email protected] Received: 5 November 2020; Accepted: 23 November 2020; Published: 25 November 2020 Abstract: The goal of this work is to study the influence of different curing systems on the cure characteristics and performance of styrene–butadiene elastomer (SBR) filled with carbon black or nanosized silica. A multifunctional additive for rubber compounds, namely Activ8, was applied as an additional activator and accelerator to increase the efficiency of sulfur vulcanization and to reduce the content of zinc oxide elastomers cured in the presence of 2-mercaptobenzothizole or 1,3-diphenylguanidine as a primary accelerator. The influence of the curing system composition on the crosslink density and physical properties of SBR vulcanizates, such as mechanical properties, thermal stability, and resistance to thermo-oxidative aging, is also reported. Activ8 effectively supports the vulcanization of SBR compounds, especially filled with nanosized silica. It reduces the optimal vulcanization time of SBR compounds and increases the crosslink density of the vulcanizates. Moreover, vulcanizates with Activ8 exhibit higher tensile strength and better damping properties than elastomer with zinc oxide. Activ8 allows the amount of ZnO to be reduced by 40% without detrimental effects on the crosslink density and mechanical performance compared to the vulcanizates conventionally crosslinked with ZnO. This is an important ecological goal since ZnO is classified as being toxic to aquatic species.