Facile Synthesis of Stable, Water Soluble, Dendron-Coated Gold Nanoparticles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Study of the Decomposition of Some Aromatic Diazonium Hexafluorophosphate Salts
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 1-1-1964 A study of the decomposition of some aromatic diazonium hexafluorophosphate salts. William A. Redmond University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Redmond, William A., "A study of the decomposition of some aromatic diazonium hexafluorophosphate salts." (1964). Electronic Theses and Dissertations. 6038. https://scholar.uwindsor.ca/etd/6038 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. A STUDY OF THE DECOMPOSITION OF SOME AROMATIC DIAZONIUM HEXAFLUOROPHOSPHATE SALTS BY WILLIAM A. REDMOND A Thesis Submitted to the Faculty of Graduate Studies through' the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the University of Windsor Windsor, Ontario 1964 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. -

Preparation of 5-Bromo-2-Naphthol: the Use of a Sulfonic Acid As a Protecting and Activating Group
Molbank 2009, M602 OPEN ACCESS molbank ISSN 1422-8599 www.mdpi.com/journal/molbank Short Note Preparation of 5-Bromo-2-naphthol: The Use of a Sulfonic Acid as a Protecting and Activating Group Renata Everett, Jillian Hamilton and Christopher Abelt * Department of Chemistry, College of William and Mary, Williamsburg, VA 23187, USA * Author to whom correspondence should be addressed; E-mail: [email protected] Received: 2 June 2009 / Accepted: 25 June 2009 / Published: 29 June 2009 Abstract: The preparation of 5-bromo-2-naphthol (4) in three steps from 5-amino-2- naphthol (1) is described. A sulfonic acid group is introduced at the 1-position as an activating and protecting group for the Sandmeyer reaction. The sulfonate group allows for the use of only water and sulfuric acid as solvents. The sulfonic acid is introduced with three equivalents of sulfuric acid, and it is removed in 20% aq. sulfuric acid. Keywords: Sandmeyer reaction; protecting group; sulfonation; desulfonation 1. Introduction Regioselective synthesis of disubstituted naphthalenes can be challenging especially when the substituents are on different rings. We needed 5-bromo-2-naphthol (4) as a starting material for a multistep synthesis. This simple derivative is virtually unknown [1,2]. The most direct route to 4 is from 5-amino-2-naphthol (1) using the Sandmeyer reaction. Unfortunately, the Sandmeyer reaction fails with 1 because the hydroxyl group is too activating. Even when the hydroxyl group is protected as a methyl ether, the normal solution-phase Sandmeyer reaction employing cuprous salts is still problematic. In their preparation of 5-bromo-2-methoxynaphthalene, Dauben and co-workers resorted to pyrolysis of the diazonium ion double salt with HgBr2, but this procedure gives just a 30% yield of the bromide [3]. -

The Electrical Properties of Graphene Modified by Bromophenyl Groups
CARBON 50 (2012) 1517– 1522 Available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/carbon The electrical properties of graphene modified by bromophenyl groups derived from a diazonium compound Xiaochen Dong a, Qing Long a, Ang Wei a, Wenjing Zhang c, Lain-Jong Li c, Peng Chen b,*, Wei Huang a,* a Key Laboratory for Organic Electronics and Information Displays (KLOEID), Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications (NUPT), 9 Wenyuan Road, Nanjing 210046, China b School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637457, Singapore c Research Center for Applied Sciences, Academia Sinica, Taipei 11529, Taiwan ARTICLE INFO ABSTRACT Article history: Graphene field-effect transistors were fabricated with mechanically exfoliated single-layer Received 30 June 2011 graphene (SLG) and bilayer graphene (BLG) sheets and the functionalization effects of Accepted 15 November 2011 bromophenyl groups derived from a diazonium compound on its transfer properties were Available online 25 November 2011 explored. Spectroscopic and electrical studies reveal that the bromophenyl grafting imposes p-doping to both SLG and BLG. The modification of SLG by bromophenyl groups significantly reduces the hole carrier mobility and the saturation current in SLG transistors, suggesting an increase in both long-range impurity and short-range defect scattering. Unexpectedly, the bromophenyl group functionalization on BLG does not obviously increase both types of scattering, indicating that the BLG is relatively more resistant to charge- or defect-induced scattering. The results indicate that chemical modification is a simple approach to tailor the electrical properties of graphene sheets with different num- bers of layers. -

Mechanistic Studies Into the Reactions of Diazonium And
MECHANISTIC STUDIES INTO THE REACTIONS OF DIAZONIUM AND RELATED COMPOUNDS by CHARLES DOUGLAS MURRAY, B.Sc. Thesis presented for the degree of DOCTOR of PHILOSOPHY University of Edinburgh September 1974 GLBY TO MY PARENTS Douglas William and Isabella Seaton McKenzie Murray • • AND MY BROTHER • • William Graeme Murray I declare that this thesis is my own composition, that the work of which it is a record has been carried.out by myself, and that it has not been submitted in any previcis application for a Higher Degree. The thesis describes results of research carried out • in the Department of Chemistry, University of Edinburgh under the supervision of Professor J. I. G. Cadogan since the 1st October, • 1971, the date of my admission as a research student. The following is a statement of postgraduate courses attended during the last three years: Summer School in Mass Spectrometry, University of Sheffield, 26-30 March 1972; - Recent Developments in the Theory of Concerted Processes, Dr. A. J. Bellamy (five lectures); Organometallic Processes in Organic Chemistry, Professor P. L. Pauson (five lectures); Industrial Research and Development, Dr. B. Gravenor (five lectures); E. U. Chemistry Department Seminars (twenty-five periods). • ACKNOWLEDGEMENTS I should like to express my gratitude to Professor J. I. G. Cadogan for suggesting the topic of research and to Professor Cadogan and Dr. J. T. Sharp for their guidance and encouragement in all aspects of this work. I would also like to record my thanks and appreciation to various other members of Edinburgh University Chemistry Department: to Dr. R. M.Paton for advice and assistance related to e. -

J.Phys. Chem. C 2016
Article pubs.acs.org/JPCC Controlled Modification of Polymer Surfaces through Grafting of Calix[4]arene-Tetradiazoate Salts † ‡ ∥ ∥ ‡ Ludovic Troian-Gautier, Daniel E. Martínez-Tong, Julie Hubert, Francoiş Reniers, Michele Sferrazza, † § † Alice Mattiuzzi,*, Corinne Lagrost,*, and Ivan Jabin † Laboratoire de Chimie Organique, UniversitéLibre de Bruxelles (ULB), CP 160/06, 50 avenue F.D. Roosevelt, 1050 Brussels, Belgium ‡ Departement́ de Physique, Facultédes Sciences, UniversitéLibre de Bruxelles (ULB), CP 223, Campus de la Plaine, Boulevard du Triomphe, 1050 Brussels, Belgium § Institut des Sciences Chimiques de Rennes UMR n° 6226, Universitéde Rennes 1 and CNRS Equipe MaCSE, Campus de Beaulieu, Rennes 35042 Cedex, France ∥ Chimie Analytique et Chimie des Interfaces, UniversitéLibre de Bruxelles (ULB), CP 255, Campus de la Plaine, Boulevard du Triomphe, 1050 Brussels, Belgium *S Supporting Information ABSTRACT: An attractive methodology based on diazonium chemistry has been developed for the surface modification of polymers such as polypropylene (PP), polyethylene tereph- thalate (PET), and polystyrene (PS). The grafting procedure involves the in situ formation of diazoates in basic aqueous solution. The reactivity of calix[4]arene-tetradiazonium salts and a classical aryldiazonium salt was examined through comparative studies on gold and polymer surfaces. The surfaces were analyzed with a combination of techniques such as AFM, XPS, and ellipsometry. The results highlighted the fact that the calix[4]arene molecules are grafted as a robust and uniform monolayer both on gold and polymer surfaces, allowing a fine control over surface modification. Furthermore, the chemical postfunctionalization of the grafted calixarene platforms equipped with carboxylic-pendant groups was successfully performed with either an amine or an alcohol. -
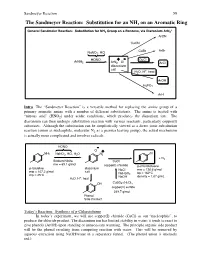
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Diazonium Ion Assay Reagents and Methods for Their Use
Patentamt Europaisches ||| || 1 1| || || || || || || || ||| ||| || (19) J European Patent Office Office europeen des brevets (11) EP 0 91 8 220 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication:ation: (51) |nt. ci.6: G01N 33/72, C07C 245/20 26.05.1999 Bulletin 1999/21 (21) Application number: 98121814.2 (22) Dateof filing: 17.11.1998 (84) Designated Contracting States: • Gordon, Tracey E. AT BE CH CY DE DK ES Fl FR GB GR IE IT LI LU Nobelsville, Indiana 46060 (US) MCNLPTSE • Bournique, Jennifer S. Designated Extension States: Indianapolis, Indiana 46239-9500 (US) AL LT LV MK RO SI • Walker, Sharanpal K. Indianapolis, Indiana 46620 (US) (30) Priority: 21.11.1997 US 975796 (74) Representative: (71) Applicant: Schwarz, Ralf, Dr. BOEHRINGER MANNHEIM CORPORATION Roche Diagnostics GmbH, Indianapolis, Indiana 46250-0528 (US) Patentabteilung, Sandhofer Strasse 116 (72) Inventors: 68305 Mannheim (DE) • Gnezda, Matthew F. Fishers, Indiana 46038 (US) (54) Diazonium ion assay reagents and methods for their use (57) Diazonium ions which are useful as reagents ducibly detected, and optionally correlated with the for the assay of bilirubin content in a sample, such as a presence or absence of any of a variety of diseases or body fluid sample, are provided. In one preferred disorders of organs such as the liver, gall bladder or embodiment, 2-methyl-3-nitroaniline diazonium ion is intestines. Using the assays, a wide range of different provided, having the structure: body fluid samples, such as urine, plasma or serum samples may be tested, and the interference in the assay from other components of the sample may be +N=N minimized. -

Molecular Transport Properties Through Carbon Nanotube Membranes
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2007 MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES Mainak Majumder University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Majumder, Mainak, "MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES" (2007). University of Kentucky Doctoral Dissertations. 557. https://uknowledge.uky.edu/gradschool_diss/557 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Mainak Majumder The Graduate School University of Kentucky 2007 MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Engineering at the University of Kentucky By Mainak Majumder Lexington, Kentucky Director: Dr. Bruce J. Hinds, William Bryan Professor of Materials Engineering Lexington, Kentucky 2007 Copyright © Mainak Majumder 2007 ABSTRACT OF DISSERTATION MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES Molecular transport through hollow cores of crystalline carbon nanotubes (CNTs) are of considerable interest from the fundamental and application point of view. This dissertation focuses on understanding molecular transport through a membrane platform consisting of open ended CNTs with ~ 7 nm core diameter and ~ 1010 CNTs/cm2 encapsulated in an inert polymer matrix. While ionic diffusion through the membrane is close to bulk diffusion expectations, gases and liquids were respectively observed to be transported ~ 10 times faster than Knudsen diffusion and ~ 10000-100000 times faster than hydrodynamic flow predictions. -

Chemistry Unit 5
EDEXCEL Advanced Level Edexcel IAL Chemistry Unit 2 Summary© HASAN SAYGINEL HS Redox and chemistry of the transition metals 1 Application of redox equilibria 1 TERMINOLOGY Oxidation number: The oxidation numbers of the elements in a compound are the charges they would have if the electrons in each bond of the molecule or ion belonged to the more electronegative element. The oxidation number of an ion is the number of electrons that have been removed (cation) or added (anion). The oxidation number of an uncombined element or compound is zero. • The oxidation number for an atom of any free (uncombined) element is ZERO. • The oxidation number of an element in self-combination is always ZERO. • In most hydrogen containing compounds, oxidation number of hydrogen is + 1.(Exception is when H combines with alkali metals or alkaline earth to form hydrides of metals such as: NaH, LiH, CaH2. Then, the oxidation number of H is -1). • In compounds involving the alkali metals, the elements are assigned oxidation number of +1. • In compounds involving the alkaline earth metals, the elements are assigned oxidation number of +2. • Oxygen is usually assigned an oxidation number of -2 for oxides. It has an oxidation number of -1 in peroxides (H2O2). • Fluorine always has oxidation number of -1 in compounds. The other elements in that group are usually -1 in compounds with elements of low electronegativity. • The sum of oxidation numbers of all the atoms in the formula for a neutral compound is ZERO. • The sum of oxidation numbers of an ion or complex ion is the same as the charge on that ion. -

Carcinogenesis of 4-(Hydroxymethyl)Benzenediazonium Ion (Tetrafluoroborate) of Agaricus Bisporus1
[CANCER RESEARCH 41, 2444-2449, June 1981] 0008-5472/81 /0041-0000 $02.00 Carcinogenesis of 4-(Hydroxymethyl)benzenediazonium Ion (Tetrafluoroborate) of Agaricus bisporus1 Bela Toth, Kasinath Patii, and Hwan-Soo Jae Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, Nebraska 68105 ABSTRACT MATERIALS AND METHODS 4-(Hydroxymethyl)benzenediazonium tetrafluoroborate was Swiss albino mice from the colony randomly bred by us since administered as 26 weekly s.c. injections of 50 /ig/g body 1951 were used. They were housed in plastic cages with weight to randomly bred Swiss mice. In addition, as a solvent granular cellulose bedding, separated according to sex in control, sodium tetrafluoroborate was given as 26 weekly s.c. groups of 5, and given Wayne Lab-Blox diet in regular pellets injections at 25 fig/g body weight in 0.9% NaCI solution to (Allied Mills, Inc., Chicago, III.) and tap water ad libitum. another group of mice. The 4-(hydroxymethyl)benzenedi- The chemicals used were HMBD (Chart 1) (M.W. 221.96; azonium tetrofluoroborate treatment induced tumors in the m.p. 61-62°; purity, 95%) and STB (M.W. 103.79; m.p. 384° subcutis and skin in incidences of 20 and 12%, respectively; with decomposition; Alfa Division, Danvers, Mass.). HMBD was while in the solvent sodium tetrafluoroborate-injected mice, the synthesized in this laboratory in the following way. corresponding tumor incidences were 6 and 0%, respectively. A 1.75-g (1.5 mmol) sample of nitrosyl tetrafluoroborate was Histopathologically, the tumors were classified as a fibroma, suspended in 15 ml of ethyl acetate maintained at 0°. -

Understanding a Mild, Metal Free Reduction Method for the Large Scale Synthesis of Hydrazines
Tetrahedron 67 (2011) 10296e10303 Contents lists available at SciVerse ScienceDirect Tetrahedron journal homepage: www.elsevier.com/locate/tet Piecing together the puzzle: understanding a mild, metal free reduction method for the large scale synthesis of hydrazines Duncan L. Browne *, Ian R. Baxendale, Steven V. Ley Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, United Kingdom article info abstract Article history: A key intermediate for the synthesis of hydrazines via a mild, metal free reduction of diazonium salts has Received 8 August 2011 been isolated and characterized by X-ray analysis. The presence of this intermediate is general, as Received in revised form 29 September 2011 demonstrated by the preparation of a number of analogues. A discussion of the mechanism and potential Accepted 30 September 2011 benefits of such a process are also described. Available online 12 October 2011 Ó 2011 Elsevier Ltd. All rights reserved. Dedicated to Professor Stork on the occasion of his 90th birthday Keywords: Hydrazines Vitamin C Ascorbic acid Diazonium salts X-ray 1. Introduction dicarbonyls (or equivalent surrogates) to provide pyrazoles, or by a Fischer indole synthesis employing the appropriate aldehyde or Of the top 200 selling pharmaceuticals in recent years, 20% of ketone (Fig. 1). Such synthesis methods are, however, not without the five-membered heterocyclic ring systems present are pyrazoles inherent problems, including relatively poor control over regiose- or indoles (Fig. 1).1 These motifs are usually derived from mono- lectivity. The requirement for the use of strong acid catalysis and substituted hydrazines either by condensation with 1,3- forcing reaction conditions is also of note particularly for the Fig. -

US3215529.Pdf
Nov. 2, 1965 R. M. LINDORUIST ETAL 3,215,529 COLOR PHOTOGRAPHIC MATERIAL Filed July 13, 1960 NE" 4. 2 8 22 INVENTORS /72aata 7/24 MMVAGeMM67 Az a ya //z Zay /777 Hur UA Cap/rcaya, ow "4e) 24%isATTORNEYS 3,215,529 United States Patent Office Patented Nov. 2, 1965 2 3,215,529 base is designated by the numeral 2, same being coated COLOR PHOTOGRAPHC MATERIAL on one side with a photosensitive layer 4 containing the Robert M. Lindquist, Lexington, Ky., Floyd T. Neth, leuconitrile color former. Typically, layer 4 may com Pensacola, Fla., and Arthur J. Critchlow, San Jose, prise a color former such as hydroxyethylate p-rosaniline Calif., assignors of one-half to Kalvar Corporation, leuconitrile, which gives a magenta image, in gelatin, New Orleans, La., a corporation of Louisiana, and of The superimposed coating 6 comprises the photosensitive one-half to International Business Machines Corpora layer which forms light scattering centers upon ex tion, San Jose, Calif., a corporation of New York posure to ultraviolet light above 3500A., and heat de Filed July 18, 1960, Ser. No. 43,374 velopment. Typically, this may comprise a suspension 7 Claims. (C. 96-27) 10 of light sensitive diazonium compound in a thermoplastic The present invention relates to a novel photographic polymer carrier such as "Saran' (vinylidene chloride color material. More particularly, the invention is con acrylonitrile copolymer) as described in Serial No. cerned with color photographic film comprising a plu 623,050. The other side of base 2 is coated with the rality of integral layers of different photosensitivity photosensitive layer 8 containing a different leuconitrile capable of dry processing.