A Study of the Decomposition of Some Aromatic Diazonium Hexafluorophosphate Salts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Microflex Gloves Chemical Compatibility Chart
1 1 1 2 2 3 1 CAUTION (LATEX): This product contains natural rubber 2 CAUTION (NITRILE: MEDICAL GRADE): Components used 3 CAUTION (NITRILE: NON-MEDICAL GRADE)): These latex (latex) which may cause allergic reactions. Safe use in making these gloves may cause allergic reactions in gloves are for non-medical use only. They may NOT be of this glove by or on latex sensitized individuals has not some users. Follow your institution’s policies for use. worn for barrier protection in medical or healthcare been established. applications. Please select other gloves for these applications. Components used in making these gloves may cause allergic reactions in some users. Follow your institution’s policies for use. For single use only. NeoPro® Chemicals NeoPro®EC Ethanol ■NBT Ethanolamine (99%) ■NBT Ether ■2 Ethidium bromide (1%) ■NBT Ethyl acetate ■1 Formaldehyde (37%) ■NBT Formamide ■NBT Gluteraldehyde (50%) ■NBT Test Method Description: The test method uses analytical Guanidine hydrochloride ■NBT equipment to determine the concentration of and the time at which (50% ■0 the challenge chemical permeates through the glove film. The Hydrochloric acid ) liquid challenge chemical is collected in a liquid miscible chemical Isopropanol ■NBT (collection media). Data is collected in three separate cells; each cell Methanol ■NBT is compared to a blank cell which uses the same collection media as both the challenge and Methyl ethyl ketone ■0 collection chemical. Methyl methacrylate (33%) ■0 Cautionary Information: These glove recommendations are offered as a guide and for reference Nitric acid (50%) ■NBT purposes only. The barrier properties of each glove type may be affected by differences in material Periodic acid (50%) ■NBT thickness, chemical concentration, temperature, and length of exposure to chemicals. -

Preparation of 5-Bromo-2-Naphthol: the Use of a Sulfonic Acid As a Protecting and Activating Group
Molbank 2009, M602 OPEN ACCESS molbank ISSN 1422-8599 www.mdpi.com/journal/molbank Short Note Preparation of 5-Bromo-2-naphthol: The Use of a Sulfonic Acid as a Protecting and Activating Group Renata Everett, Jillian Hamilton and Christopher Abelt * Department of Chemistry, College of William and Mary, Williamsburg, VA 23187, USA * Author to whom correspondence should be addressed; E-mail: [email protected] Received: 2 June 2009 / Accepted: 25 June 2009 / Published: 29 June 2009 Abstract: The preparation of 5-bromo-2-naphthol (4) in three steps from 5-amino-2- naphthol (1) is described. A sulfonic acid group is introduced at the 1-position as an activating and protecting group for the Sandmeyer reaction. The sulfonate group allows for the use of only water and sulfuric acid as solvents. The sulfonic acid is introduced with three equivalents of sulfuric acid, and it is removed in 20% aq. sulfuric acid. Keywords: Sandmeyer reaction; protecting group; sulfonation; desulfonation 1. Introduction Regioselective synthesis of disubstituted naphthalenes can be challenging especially when the substituents are on different rings. We needed 5-bromo-2-naphthol (4) as a starting material for a multistep synthesis. This simple derivative is virtually unknown [1,2]. The most direct route to 4 is from 5-amino-2-naphthol (1) using the Sandmeyer reaction. Unfortunately, the Sandmeyer reaction fails with 1 because the hydroxyl group is too activating. Even when the hydroxyl group is protected as a methyl ether, the normal solution-phase Sandmeyer reaction employing cuprous salts is still problematic. In their preparation of 5-bromo-2-methoxynaphthalene, Dauben and co-workers resorted to pyrolysis of the diazonium ion double salt with HgBr2, but this procedure gives just a 30% yield of the bromide [3]. -

The Electrical Properties of Graphene Modified by Bromophenyl Groups
CARBON 50 (2012) 1517– 1522 Available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/carbon The electrical properties of graphene modified by bromophenyl groups derived from a diazonium compound Xiaochen Dong a, Qing Long a, Ang Wei a, Wenjing Zhang c, Lain-Jong Li c, Peng Chen b,*, Wei Huang a,* a Key Laboratory for Organic Electronics and Information Displays (KLOEID), Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications (NUPT), 9 Wenyuan Road, Nanjing 210046, China b School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637457, Singapore c Research Center for Applied Sciences, Academia Sinica, Taipei 11529, Taiwan ARTICLE INFO ABSTRACT Article history: Graphene field-effect transistors were fabricated with mechanically exfoliated single-layer Received 30 June 2011 graphene (SLG) and bilayer graphene (BLG) sheets and the functionalization effects of Accepted 15 November 2011 bromophenyl groups derived from a diazonium compound on its transfer properties were Available online 25 November 2011 explored. Spectroscopic and electrical studies reveal that the bromophenyl grafting imposes p-doping to both SLG and BLG. The modification of SLG by bromophenyl groups significantly reduces the hole carrier mobility and the saturation current in SLG transistors, suggesting an increase in both long-range impurity and short-range defect scattering. Unexpectedly, the bromophenyl group functionalization on BLG does not obviously increase both types of scattering, indicating that the BLG is relatively more resistant to charge- or defect-induced scattering. The results indicate that chemical modification is a simple approach to tailor the electrical properties of graphene sheets with different num- bers of layers. -

Mechanistic Studies Into the Reactions of Diazonium And
MECHANISTIC STUDIES INTO THE REACTIONS OF DIAZONIUM AND RELATED COMPOUNDS by CHARLES DOUGLAS MURRAY, B.Sc. Thesis presented for the degree of DOCTOR of PHILOSOPHY University of Edinburgh September 1974 GLBY TO MY PARENTS Douglas William and Isabella Seaton McKenzie Murray • • AND MY BROTHER • • William Graeme Murray I declare that this thesis is my own composition, that the work of which it is a record has been carried.out by myself, and that it has not been submitted in any previcis application for a Higher Degree. The thesis describes results of research carried out • in the Department of Chemistry, University of Edinburgh under the supervision of Professor J. I. G. Cadogan since the 1st October, • 1971, the date of my admission as a research student. The following is a statement of postgraduate courses attended during the last three years: Summer School in Mass Spectrometry, University of Sheffield, 26-30 March 1972; - Recent Developments in the Theory of Concerted Processes, Dr. A. J. Bellamy (five lectures); Organometallic Processes in Organic Chemistry, Professor P. L. Pauson (five lectures); Industrial Research and Development, Dr. B. Gravenor (five lectures); E. U. Chemistry Department Seminars (twenty-five periods). • ACKNOWLEDGEMENTS I should like to express my gratitude to Professor J. I. G. Cadogan for suggesting the topic of research and to Professor Cadogan and Dr. J. T. Sharp for their guidance and encouragement in all aspects of this work. I would also like to record my thanks and appreciation to various other members of Edinburgh University Chemistry Department: to Dr. R. M.Paton for advice and assistance related to e. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title THE USE OF BASIC POLYMER SORBENTS FOR THE RECOVERY OF ACETIC ACID FROM DILUTE AQUEOUS SOLUTION Permalink https://escholarship.org/uc/item/0qs29271 Authors Garcia, A.A. King, C . J . Publication Date 1988 eScholarship.org Powered by the California Digital Library University of California LBL-24543 C'_~ ITll Lawrence Berkeley Laboratory iii:~ UNIVERSITY OF CALIFORNIA APPLIED SCIENCE -. ~ ., .. I DIVISION. •-, l:: '-' 1:: v L _ LAWRENCE EJr.:ov~L''"V_, .nc: c' LABORATORY APR 1 9 1988 LIBRARY AND The Use of Basic Polymer Sorbents DOCUMENTS SECTION for the Recovery of Acetic Acid from Dilute Aqueous Solution A.A. Garcia and C.J. King January 1988 '· . J I • .I APPLIED SCIENCE DIVISION Prepared for the U.S. Department of Energy under Contract DE-AC03-76SF00098 DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of Califomia. -

J.Phys. Chem. C 2016
Article pubs.acs.org/JPCC Controlled Modification of Polymer Surfaces through Grafting of Calix[4]arene-Tetradiazoate Salts † ‡ ∥ ∥ ‡ Ludovic Troian-Gautier, Daniel E. Martínez-Tong, Julie Hubert, Francoiş Reniers, Michele Sferrazza, † § † Alice Mattiuzzi,*, Corinne Lagrost,*, and Ivan Jabin † Laboratoire de Chimie Organique, UniversitéLibre de Bruxelles (ULB), CP 160/06, 50 avenue F.D. Roosevelt, 1050 Brussels, Belgium ‡ Departement́ de Physique, Facultédes Sciences, UniversitéLibre de Bruxelles (ULB), CP 223, Campus de la Plaine, Boulevard du Triomphe, 1050 Brussels, Belgium § Institut des Sciences Chimiques de Rennes UMR n° 6226, Universitéde Rennes 1 and CNRS Equipe MaCSE, Campus de Beaulieu, Rennes 35042 Cedex, France ∥ Chimie Analytique et Chimie des Interfaces, UniversitéLibre de Bruxelles (ULB), CP 255, Campus de la Plaine, Boulevard du Triomphe, 1050 Brussels, Belgium *S Supporting Information ABSTRACT: An attractive methodology based on diazonium chemistry has been developed for the surface modification of polymers such as polypropylene (PP), polyethylene tereph- thalate (PET), and polystyrene (PS). The grafting procedure involves the in situ formation of diazoates in basic aqueous solution. The reactivity of calix[4]arene-tetradiazonium salts and a classical aryldiazonium salt was examined through comparative studies on gold and polymer surfaces. The surfaces were analyzed with a combination of techniques such as AFM, XPS, and ellipsometry. The results highlighted the fact that the calix[4]arene molecules are grafted as a robust and uniform monolayer both on gold and polymer surfaces, allowing a fine control over surface modification. Furthermore, the chemical postfunctionalization of the grafted calixarene platforms equipped with carboxylic-pendant groups was successfully performed with either an amine or an alcohol. -

Annex XV Dossier PROPOSAL for IDENTIFICATION of A
ANNEX XV – IDENTIFICATION OF N,N-DIMETHYLACETAMIDE (DMAC) AS SVHC Annex XV dossier PROPOSAL FOR IDENTIFICATION OF A SUBSTANCE AS A CATEGORY 1A OR 1B CMR, PBT, vPvB OR A SUBSTANCE OF AN EQUIVALENT LEVEL OF CONCERN Substance Name(s): N,N-Dimethylacetamide (DMAC) EC Number(s): 204-826-4 CAS Number(s): 127-19-5 Submitted by: European Chemicals Agency at the request of the European Commission Version: August 2011 PUBLIC VERSION: This version does not include the confidential annexes referred to in Parts I and II. 1 ANNEX XV – IDENTIFICATION OF N,N-DIMETHYLACETAMIDE (DMAC) AS SVHC CONTENTS ABBREVIATIONS AND ACRONYMS.....................................................................................................................6 PART I..........................................................................................................................................................................9 JUSTIFICATION .........................................................................................................................................................9 1 IDENTITY OF THE SUBSTANCE AND PHYSICAL AND CHEMICAL PROPERTIES .................................9 1.1 Name and other identifiers of the substance ...................................................................................................9 1.2 Composition of the substance.........................................................................................................................10 1.3 Physico-chemical properties...........................................................................................................................11 -
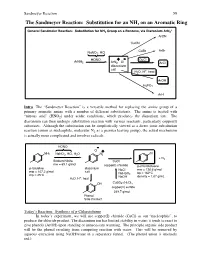
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Dissociation Constants of Organic Acids and Bases
DISSOCIATION CONSTANTS OF ORGANIC ACIDS AND BASES This table lists the dissociation (ionization) constants of over pKa + pKb = pKwater = 14.00 (at 25°C) 1070 organic acids, bases, and amphoteric compounds. All data apply to dilute aqueous solutions and are presented as values of Compounds are listed by molecular formula in Hill order. pKa, which is defined as the negative of the logarithm of the equi- librium constant K for the reaction a References HA H+ + A- 1. Perrin, D. D., Dissociation Constants of Organic Bases in Aqueous i.e., Solution, Butterworths, London, 1965; Supplement, 1972. 2. Serjeant, E. P., and Dempsey, B., Ionization Constants of Organic Acids + - Ka = [H ][A ]/[HA] in Aqueous Solution, Pergamon, Oxford, 1979. 3. Albert, A., “Ionization Constants of Heterocyclic Substances”, in where [H+], etc. represent the concentrations of the respective Katritzky, A. R., Ed., Physical Methods in Heterocyclic Chemistry, - species in mol/L. It follows that pKa = pH + log[HA] – log[A ], so Academic Press, New York, 1963. 4. Sober, H.A., Ed., CRC Handbook of Biochemistry, CRC Press, Boca that a solution with 50% dissociation has pH equal to the pKa of the acid. Raton, FL, 1968. 5. Perrin, D. D., Dempsey, B., and Serjeant, E. P., pK Prediction for Data for bases are presented as pK values for the conjugate acid, a a Organic Acids and Bases, Chapman and Hall, London, 1981. i.e., for the reaction 6. Albert, A., and Serjeant, E. P., The Determination of Ionization + + Constants, Third Edition, Chapman and Hall, London, 1984. BH H + B 7. Budavari, S., Ed., The Merck Index, Twelth Edition, Merck & Co., Whitehouse Station, NJ, 1996. -

Diazonium Ion Assay Reagents and Methods for Their Use
Patentamt Europaisches ||| || 1 1| || || || || || || || ||| ||| || (19) J European Patent Office Office europeen des brevets (11) EP 0 91 8 220 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication:ation: (51) |nt. ci.6: G01N 33/72, C07C 245/20 26.05.1999 Bulletin 1999/21 (21) Application number: 98121814.2 (22) Dateof filing: 17.11.1998 (84) Designated Contracting States: • Gordon, Tracey E. AT BE CH CY DE DK ES Fl FR GB GR IE IT LI LU Nobelsville, Indiana 46060 (US) MCNLPTSE • Bournique, Jennifer S. Designated Extension States: Indianapolis, Indiana 46239-9500 (US) AL LT LV MK RO SI • Walker, Sharanpal K. Indianapolis, Indiana 46620 (US) (30) Priority: 21.11.1997 US 975796 (74) Representative: (71) Applicant: Schwarz, Ralf, Dr. BOEHRINGER MANNHEIM CORPORATION Roche Diagnostics GmbH, Indianapolis, Indiana 46250-0528 (US) Patentabteilung, Sandhofer Strasse 116 (72) Inventors: 68305 Mannheim (DE) • Gnezda, Matthew F. Fishers, Indiana 46038 (US) (54) Diazonium ion assay reagents and methods for their use (57) Diazonium ions which are useful as reagents ducibly detected, and optionally correlated with the for the assay of bilirubin content in a sample, such as a presence or absence of any of a variety of diseases or body fluid sample, are provided. In one preferred disorders of organs such as the liver, gall bladder or embodiment, 2-methyl-3-nitroaniline diazonium ion is intestines. Using the assays, a wide range of different provided, having the structure: body fluid samples, such as urine, plasma or serum samples may be tested, and the interference in the assay from other components of the sample may be +N=N minimized. -

Molecular Transport Properties Through Carbon Nanotube Membranes
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2007 MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES Mainak Majumder University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Majumder, Mainak, "MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES" (2007). University of Kentucky Doctoral Dissertations. 557. https://uknowledge.uky.edu/gradschool_diss/557 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Mainak Majumder The Graduate School University of Kentucky 2007 MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the College of Engineering at the University of Kentucky By Mainak Majumder Lexington, Kentucky Director: Dr. Bruce J. Hinds, William Bryan Professor of Materials Engineering Lexington, Kentucky 2007 Copyright © Mainak Majumder 2007 ABSTRACT OF DISSERTATION MOLECULAR TRANSPORT PROPERTIES THROUGH CARBON NANOTUBE MEMBRANES Molecular transport through hollow cores of crystalline carbon nanotubes (CNTs) are of considerable interest from the fundamental and application point of view. This dissertation focuses on understanding molecular transport through a membrane platform consisting of open ended CNTs with ~ 7 nm core diameter and ~ 1010 CNTs/cm2 encapsulated in an inert polymer matrix. While ionic diffusion through the membrane is close to bulk diffusion expectations, gases and liquids were respectively observed to be transported ~ 10 times faster than Knudsen diffusion and ~ 10000-100000 times faster than hydrodynamic flow predictions. -

Chemistry Unit 5
EDEXCEL Advanced Level Edexcel IAL Chemistry Unit 2 Summary© HASAN SAYGINEL HS Redox and chemistry of the transition metals 1 Application of redox equilibria 1 TERMINOLOGY Oxidation number: The oxidation numbers of the elements in a compound are the charges they would have if the electrons in each bond of the molecule or ion belonged to the more electronegative element. The oxidation number of an ion is the number of electrons that have been removed (cation) or added (anion). The oxidation number of an uncombined element or compound is zero. • The oxidation number for an atom of any free (uncombined) element is ZERO. • The oxidation number of an element in self-combination is always ZERO. • In most hydrogen containing compounds, oxidation number of hydrogen is + 1.(Exception is when H combines with alkali metals or alkaline earth to form hydrides of metals such as: NaH, LiH, CaH2. Then, the oxidation number of H is -1). • In compounds involving the alkali metals, the elements are assigned oxidation number of +1. • In compounds involving the alkaline earth metals, the elements are assigned oxidation number of +2. • Oxygen is usually assigned an oxidation number of -2 for oxides. It has an oxidation number of -1 in peroxides (H2O2). • Fluorine always has oxidation number of -1 in compounds. The other elements in that group are usually -1 in compounds with elements of low electronegativity. • The sum of oxidation numbers of all the atoms in the formula for a neutral compound is ZERO. • The sum of oxidation numbers of an ion or complex ion is the same as the charge on that ion.