Bayesian and Likelihood Placement of Fossils on Phylogenies from Quantitative
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Full Article in PDF Format
A new marine vertebrate assemblage from the Late Neogene Purisima Formation in Central California, part II: Pinnipeds and Cetaceans Robert W. BOESSENECKER Department of Geology, University of Otago, 360 Leith Walk, P.O. Box 56, Dunedin, 9054 (New Zealand) and Department of Earth Sciences, Montana State University 200 Traphagen Hall, Bozeman, MT, 59715 (USA) and University of California Museum of Paleontology 1101 Valley Life Sciences Building, Berkeley, CA, 94720 (USA) [email protected] Boessenecker R. W. 2013. — A new marine vertebrate assemblage from the Late Neogene Purisima Formation in Central California, part II: Pinnipeds and Cetaceans. Geodiversitas 35 (4): 815-940. http://dx.doi.org/g2013n4a5 ABSTRACT e newly discovered Upper Miocene to Upper Pliocene San Gregorio assem- blage of the Purisima Formation in Central California has yielded a diverse collection of 34 marine vertebrate taxa, including eight sharks, two bony fish, three marine birds (described in a previous study), and 21 marine mammals. Pinnipeds include the walrus Dusignathus sp., cf. D. seftoni, the fur seal Cal- lorhinus sp., cf. C. gilmorei, and indeterminate otariid bones. Baleen whales include dwarf mysticetes (Herpetocetus bramblei Whitmore & Barnes, 2008, Herpetocetus sp.), two right whales (cf. Eubalaena sp. 1, cf. Eubalaena sp. 2), at least three balaenopterids (“Balaenoptera” cortesi “var.” portisi Sacco, 1890, cf. Balaenoptera, Balaenopteridae gen. et sp. indet.) and a new species of rorqual (Balaenoptera bertae n. sp.) that exhibits a number of derived features that place it within the genus Balaenoptera. is new species of Balaenoptera is relatively small (estimated 61 cm bizygomatic width) and exhibits a comparatively nar- row vertex, an obliquely (but precipitously) sloping frontal adjacent to vertex, anteriorly directed and short zygomatic processes, and squamosal creases. -

Pinniped Evolution and Puijila Darwini
A pP pP eE nN dD iI xX AE Pinniped Evolution and Puijila darwini Pinnipeds are carnivorous marine mammals that fossils, evolution scientists have not found any defini- have “finned back feet,” similar to the fins used by a tive fossils showing a land mammal evolving into a scuba diver. The Latin-derived word “pinniped” seal, sea lion or walrus. literally means “finned-foot.” Pinnipeds include three Canadian paleobiologist and professor, Dr. Natalia groups of mammals living today; namely, sea lions, Rybczynski of the Canadian Museum of Nature, wrote seals, and walruses. this candid assessment in 2009: The “fossil evidence of By 2007, when the first edition of this book was the morphological steps leading from a terrestrial published, scientists had discovered 20,000 fossil ancestor to the modern marine forms has been weak or pinnipeds. (See Appendix A.) Despite this plethora of contentious.” 1 All three types of pinnipeds Sea lion living today, sea lions (left), walruses (bottom left), and seals (below), have finned back feet, the telltale sign of a pinniped. Seal Walrus Appendix E: Puijila Adarwini p p e n d i x 239 A P P E N D I X EA Enaliarctos—The Oldest Pinniped Enaliarctos, the oldest fossil pinniped, looks like a between a terrestrial ancestor and the appearance of sea lion, and not a missing link. 2 (See photos below.) flippered pinnipeds. Indeed, most studies of pinniped Dr. Natalia Rybczynski highlights this missing link relationships and evolution do not consider the critical problem—the absence of fossils from a land mammal -

From the Late Miocene-Early Pliocene (Hemphillian) of California
Bull. Fla. Mus. Nat. Hist. (2005) 45(4): 379-411 379 NEW SKELETAL MATERIAL OF THALASSOLEON (OTARIIDAE: PINNIPEDIA) FROM THE LATE MIOCENE-EARLY PLIOCENE (HEMPHILLIAN) OF CALIFORNIA Thomas A. Deméré1 and Annalisa Berta2 New crania, dentitions, and postcrania of the fossil otariid Thalassoleon mexicanus are described from the latest Miocene–early Pliocene Capistrano Formation of southern California. Previous morphological evidence for age variation and sexual dimorphism in this taxon is confirmed. Analysis of the dentition and postcrania of Thalassoleon mexicanus provides evidence of adaptations for pierce feeding, ambulatory terrestrial locomotion, and forelimb swimming in this basal otariid pinniped. Cladistic analysis supports recognition of Thalassoleon as monophyletic and distinct from other basal otariids (i.e., Pithanotaria, Hydrarctos, and Callorhinus). Re-evaluation of the status of Thalassoleon supports recognition of two species, Thalassoleon mexicanus and Thalassoleon macnallyae, distributed in the eastern North Pacific. Recognition of a third species, Thalassoleon inouei from the western North Pacific, is questioned. Key Words: Otariidae; pinniped; systematics; anatomy; Miocene; California INTRODUCTION perate, with a very limited number of recovered speci- Otariid pinnipeds are a conspicuous element of the ex- mens available for study. The earliest otariid, tant marine mammal assemblage of the world’s oceans. Pithanotaria starri Kellogg 1925, is known from the Members of this group inhabit the North and South Pa- early late Miocene (Tortonian Stage equivalent) and is cific Ocean, as well as portions of the southern Indian based on a few poorly preserved fossils from Califor- and Atlantic oceans and nearly the entire Southern nia. The holotype is an impression in diatomite of a Ocean. -

Feeding Kinematics and Performance Of
© 2015. Published by The Company of Biologists Ltd | Journal of Experimental Biology (2015) 218, 3229-3240 doi:10.1242/jeb.126573 RESEARCH ARTICLE Feeding kinematics and performance of basal otariid pinnipeds, Steller sea lions and northern fur seals: implications for the evolution of mammalian feeding Christopher D. Marshall1,2,*, David A. S. Rosen3 and Andrew W. Trites3 ABSTRACT pierce or raptorial biting (hereafter referred to as biting), grip-and- Feeding performance studies can address questions relevant to tear, inertial suction (hereafter referred to as suction) and filter feeding ecology and evolution. Our current understanding of feeding feeding (Adam and Berta, 2002). Among otariids, biting, grip- mechanisms for aquatic mammals is poor. Therefore, we and-tear and suction feeding modes are thought to be the most characterized the feeding kinematics and performance of five common. Only one otariid (Antarctic fur seals, Arctocephalus Steller sea lions (Eumetopias jubatus) and six northern fur seals gazella) is known to use filter feeding (Riedman, 1990; Adam and (Callorhinus ursinus). We tested the hypotheses that both species Berta, 2002). Biting is considered to be the ancestral feeding use suction as their primary feeding mode, and that rapid jaw opening condition of basal aquatic vertebrates, as well as the terrestrial was related to suction generation. Steller sea lions used suction as ancestors of pinnipeds (Adam and Berta, 2002; Berta et al., 2006). their primary feeding mode, but also used a biting feeding mode. In Morphological evidence from Puijila, while not considered a contrast, northern fur seals only used a biting feeding mode. direct ancestor to pinnipeds (Kelley and Pyenson, 2015), suggests Kinematic profiles of Steller sea lions were all indicative of suction that the ancestral biting mode was still in use as mammals feeding (i.e. -

Early Evolution of Sexual Dimorphism and Polygyny in Pinnipedia
[The final published version of this article is available online. Please check the final publication record for the latest revisions of this article: Cullen, T. M., Fraser, D., Rybczynski, N. and Schröder-Adams, C. (2014), Early evolution of sexual dimorphism and polygyny in Pinnipedia. Evolution, 68: 1469–1484. doi:10.1111/evo.12360] Early evolution of sexual dimorphism and polygyny in Pinnipedia Thomas M. Cullen1,*, Danielle Fraser2, Natalia Rybczynski3, Claudia Schröder-Adams1 1Department of Earth Sciences, Carleton University, 1125 Colonel By Drive, Ottawa, Ontario K1S 5B6, Canada 2Department of Biology, Carleton University, 1125 Colonel By Drive, Ottawa, Ontario K1S 5B6, Canada 3Palaeobiology, Canadian Museum of Nature, PO Box 3443 Stn ‘D’, Ottawa, Ontario K1P 6P4, Canada Key Words: Fossil; Mating Systems; Coevolution, Miocene; Phocidae; Otariidae; Reproductive Strategies Total words (not including figure captions, literature cited): 6977 Tables: 0. Figures: 8 All data included as supplementary information T. M. Cullen et al. !1 * Correspondence and requests for materials should be addressed to T.M.C. (current address: [email protected], Department of Ecology and Evolutionary Biology, University of Toronto, 25 Willcocks Street, Toronto, Ontario M5S 3B2, Canada). Running Header: Cullen et al. — Evolution of sexual dimorphism in pinnipeds Abstract Sexual selection is one of the earliest areas of interest in evolutionary biology. And yet, the evolutionary history of sexually dimorphic traits remains poorly characterized for most vertebrate lineages. Here we report on evidence for the early evolution of dimorphism within a model mammal group, the pinnipeds. Pinnipeds show a range of sexual dimorphism and mating systems that span the extremes of modern mammals, from monomorphic taxa with isolated and dispersed mating to extreme size dimorphism with highly ordered polygynous harem systems. -

Navigating the Monk Seal Maze
Vol. 1 / No. 2 Published by IMMA Inc. December 1998 Editorial: Navigating the Monk Seal Maze – A Guide for the Layperson Bowing to popular demand, The Monachus Guardian presents yet another satirical view of monk seal conservation. This time, we travel to Northern Greece for a United Nations Environment Programme conference in the Byzantine city of Arta… International News Regional News Cover Story: Midway’s Monk Seals Editorial: Navigating the Monk Seal Maze When attempts were made to forcibly reintroduce Hawaiian monk seals to a former breeding site at Midway Atoll, all 18 translocated animals either died or disappeared in short order. More recently, a drastic reduction in human disturbance, coinciding with the closure of Midway’s Naval Base, has encouraged monk seals to recolonise the atoll of their own accord. But with ecotourism now developing on Midway, will this success story prove short-lived? In Focus: New Discoveries in Cilicia Researchers discover a hitherto unknown population of Mediterranean Monk Seals along Turkey’s Mediterranean Coast… Perspectives: The Life & Times of Q39 A diary of events surrounding the persecution of a yearling Hawaiian monk seal on Maui… Monachus Science: Antolovic, J. Mediterranean monk seal (Monachus monachus) habitat in Vis Archipelago, the Adriatic Midway’s Monk Seals Sea. Lavigne, D.M. Historical biogeography and phylogenetic relationships among modern monk seals, Monachus spp. Neves, H.C. & R. Pires. The recuperation of a monk seal pup, Monachus monachus, in the Ilhas Desertas – the conditions for its success. Letters to the Editor Recent Publications Conservation on the Desertas Islands Publishing Info Copyright © 1998 The Monachus Guardian. -

Re-Evaluation of Morphological Characters Questions Current Views of Pinniped Origins
Vestnik zoologii, 50(4): 327–354, 2016 Evolution and Phylogeny DOI 10.1515/vzoo-2016-0040 UDC 569.5:575.86 RE-EVALUATION OF MORPHOLOGICAL CHARACTERS QUESTIONS CURRENT VIEWS OF PINNIPED ORIGINS I. A. Koretsky¹, L. G. Barnes², S. J. Rahmat¹ ¹Laboratory of Evolutionary Biology, Department of Anatomy, College of Medicine, Howard University, 520 W. St. NW, Washington, DC 20059 E-mail: [email protected] ²Department of Vertebrate Paleontology, Natural History Museum of Los Angeles County, 900 Exposition Blvd., Los Angeles, CA 90007 Re-evaluation of Morphological Characters Questions Current Views of Pinniped Origins. Koretsky, I. A., Barnes, L. G., Rahmat, S. J. — Th e origin of pinnipeds has been a contentious issue, with opposite sides debating monophyly or diphyly. Th is review uses evidence from the fossil record, combined with comparative morphology, molecular and cytogenetic investigations to evaluate the evolutionary history and phylogenetic relationships of living and fossil otarioid and phocoid pinnipeds. Molecular investigations support a monophyletic origin of pinnipeds, but disregard vital morphological data. Likewise, morphological studies support diphyly, but overlook molecular analyses. Th is review will demonstrate that a monophyletic origin of pinnipeds should not be completely accepted, as is the current ideology, and a diphyletic origin remains viable due to morphological and paleobiological analyses. Critical examination of certain characters, used by supporters of pinniped monophyly, reveals diff erent polarities, variability, or simply convergence. Th e paleontological record and our morphological analysis of important characters supports a diphyletic origin of pinnipeds, with otarioids likely arising in the North Pacifi c from large, bear-like animals and phocids arising in the North Atlantic from smaller, otter-like ancestors. -

The Evolution of Feeding and Locomotion in Seals, Sea Lions, and Walruses
Of Fin and Mouth: the Evolution of Feeding and Locomotion in Seals, Sea Lions, and Walruses Peter J. Adam UCLA, Dept. Ecology & Evolutionary Biology WHAT IS A MARINE MAMMAL? • Any mammal with a QuickTime™ and a primary dependence Photo - JPEG decompressor are needed to see this picture. on the marine environment for Cetaceans existence. Sea Otter Polar Bear Sirenia Pinnipeds PROBLEMS FACING A MARINE MAMMAL: • Respiration • Insulation • Pressure • Buoyancy • Sense organs • Salinity • Locomotion • Prey capture …others… • Marine mammal research challenging: • observation difficult • animals are very large • boats/diving expensive • Much of our knowledge about marine mammals comes from strandings and exploitation OUTLINE • What are pinnipeds? • Introduction • Phylogenetics • Evolution of pinniped feeding • Evolution of pinniped locomotion • West Indian monk seal • 34 species in 18 genera and 3 families OTARIIDAE - SEA LIONS and FUR SEALS QuickTime™ and a Photo - JPEG decompressor are needed to see this picture. PHOCIDAE - HAIR or ‘TRUE’ SEALS ODOBENIDAE - WALRUSES Early Pinnipeds • Early Miocene (37 mya) - present • Cosmopolitan (N. Pacific origin) • Seal-like bodies • Amphibious Enaliarctos Phylogenies • depict evolutionary relationships • show relative timing of lineage divergences • constructed using many “polar” characters Phylogenies • useful for mapping the evolution of interesting traits: • Amniotic egg in amniotes • Feathers in birds • Hair in mammals Phylogenies • useful for identifying incidence of convergence and other interesting evolutionary phenomena: • Warm- bloodedness in birds and mammals Marine sloth (Pliocene) Marine bear (Miocene) Pinnipedia (Oligocene) Sea Otter (Miocene) Cetacea (Eocene) Desmostylia (Oligocene) Sirenia (Eocene) Thalassocnus • Marine sloth (5 spp.) • Xenarthra • Pliocene of Peru HABITS: • herbivorous (algae) • near-shore • not a strong swimmer Kolponomus • Marine bear (4 spp.) • Carnivora (Laurasiatheria) • E. -

A Survey of Cenozoic Mammal Baramins
The Proceedings of the International Conference on Creationism Volume 8 Print Reference: Pages 217-221 Article 43 2018 A Survey of Cenozoic Mammal Baramins C Thompson Core Academy of Science Todd Charles Wood Core Academy of Science Follow this and additional works at: https://digitalcommons.cedarville.edu/icc_proceedings DigitalCommons@Cedarville provides a publication platform for fully open access journals, which means that all articles are available on the Internet to all users immediately upon publication. However, the opinions and sentiments expressed by the authors of articles published in our journals do not necessarily indicate the endorsement or reflect the views of DigitalCommons@Cedarville, the Centennial Library, or Cedarville University and its employees. The authors are solely responsible for the content of their work. Please address questions to [email protected]. Browse the contents of this volume of The Proceedings of the International Conference on Creationism. Recommended Citation Thompson, C., and T.C. Wood. 2018. A survey of Cenozic mammal baramins. In Proceedings of the Eighth International Conference on Creationism, ed. J.H. Whitmore, pp. 217–221. Pittsburgh, Pennsylvania: Creation Science Fellowship. Thompson, C., and T.C. Wood. 2018. A survey of Cenozoic mammal baramins. In Proceedings of the Eighth International Conference on Creationism, ed. J.H. Whitmore, pp. 217–221, A1-A83 (appendix). Pittsburgh, Pennsylvania: Creation Science Fellowship. A SURVEY OF CENOZOIC MAMMAL BARAMINS C. Thompson, Core Academy of Science, P.O. Box 1076, Dayton, TN 37321, [email protected] Todd Charles Wood, Core Academy of Science, P.O. Box 1076, Dayton, TN 37321, [email protected] ABSTRACT To expand the sample of statistical baraminology studies, we identified 80 datasets sampled from 29 mammalian orders, from which we performed 82 separate analyses. -

Feeding Kinematics and Performance of Basal Otariid Pinnipeds, Steller Sea Lions and Northern Fur Seals
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Texas A&M Repository © 2015. Published by The Company of Biologists Ltd | Journal of Experimental Biology (2015) 218, 3229-3240 doi:10.1242/jeb.126573 RESEARCH ARTICLE Feeding kinematics and performance of basal otariid pinnipeds, Steller sea lions and northern fur seals: implications for the evolution of mammalian feeding Christopher D. Marshall1,2,*, David A. S. Rosen3 and Andrew W. Trites3 ABSTRACT pierce or raptorial biting (hereafter referred to as biting), grip-and- Feeding performance studies can address questions relevant to tear, inertial suction (hereafter referred to as suction) and filter feeding ecology and evolution. Our current understanding of feeding feeding (Adam and Berta, 2002). Among otariids, biting, grip- mechanisms for aquatic mammals is poor. Therefore, we and-tear and suction feeding modes are thought to be the most characterized the feeding kinematics and performance of five common. Only one otariid (Antarctic fur seals, Arctocephalus Steller sea lions (Eumetopias jubatus) and six northern fur seals gazella) is known to use filter feeding (Riedman, 1990; Adam and (Callorhinus ursinus). We tested the hypotheses that both species Berta, 2002). Biting is considered to be the ancestral feeding use suction as their primary feeding mode, and that rapid jaw opening condition of basal aquatic vertebrates, as well as the terrestrial was related to suction generation. Steller sea lions used suction as ancestors of pinnipeds (Adam and Berta, 2002; Berta et al., 2006). their primary feeding mode, but also used a biting feeding mode. In Morphological evidence from Puijila, while not considered a contrast, northern fur seals only used a biting feeding mode. -
Second Revision of Recovery Plan for the Hawaiian Monk Seal
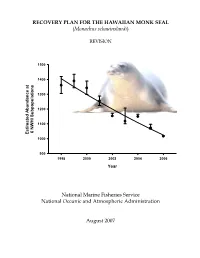
RECOVERY PLAN FOR THE HAWAIIAN MONK SEAL (Monachus schauinslandi) REVISION 1500 1400 1300 1200 1100 6 NWHI Subpopulations 6 NWHI Estimated Abundance at at Abundance Estimated 1000 900 1998 2000 2002 2004 2006 Year National Marine Fisheries Service National Oceanic and Atmospheric Administration August 2007 RECOVERY PLAN FOR THE HAWAIIAN MONK SEAL (Monachus schauinslandi) REVISION Original Version: March 1983 Prepared by National Marine Fisheries Service National Oceanic and Atmospheric Administration Approved: - __a:=-~.=...::::=:' '""""""""'""----"1_, ---,Yb"'---=--~--.f=-'--- ______ William T. Hogarth, Ph.D, Assistant Administrator for Fisheries National Oceanic and Atmospheric Administration Date August 22, 2007 DISCLAIMER Recovery plans delineate reasonable actions which the best available information indicates are necessary to recover and/or protect listed species. Plans are published by the National Marine Fisheries Service (NMFS), sometimes prepared with the assistance of recovery teams, contractors, State agencies and others. Recovery plans do not necessarily represent the views, official positions or approval of any individuals or agencies involved in the plan formulation, other than NMFS. They represent the official position of NMFS only after they have been signed by the Assistant Administrator. Recovery plans are guidance and planning documents only; identification of an action to be implemented by any public or private party does not create a legal obligation beyond existing legal requirements. Nothing in this plan should be construed as a commitment or requirement that any Federal agency obligate or pay funds in any one fiscal year in excess of appropriations made by Congress for that fiscal year in contravention of the Anti-Deficiency Act, 31 U.S.C. 1341, or any other law or regulation. -

Bayesian and Likelihood Placement of Fossils on Phylogenies from Quantitative
bioRxiv preprint doi: https://doi.org/10.1101/275446; this version posted March 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Running head: PLACING FOSSILS WITH MORPHOMETRIC DATA 2 Title: Bayesian and likelihood placement of fossils on phylogenies from quantitative 3 morphometrics 4 Caroline Parins-Fukuchi 5 Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, Michigan, 6 48109, USA. 7 E-mail: [email protected] 8 Telephone: (734) 474-7241 1 bioRxiv preprint doi: https://doi.org/10.1101/275446; this version posted March 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 9 Acknowledgements I thank James Saulsbury, Gregory Stull, Joseph Walker, and Stephen Smith for 10 helpful comments that improved the manuscript. I also greatly thank Iju Chen and Steven 11 Manchester for providing the Vitaceae measurement data, and Gregory Stull for coordinating and 12 helping to prepare the data for analysis. 13 Supplementary Data All supplemental material, including scripts, newick files, morphological and 14 molecular alignments used in this study are available as a GitHub repository 15 (https://github.com/carolinetomo/fossil_placement_tests). 2 bioRxiv preprint doi: https://doi.org/10.1101/275446; this version posted March 5, 2018.