Tmsotf-Promoted Sulfinylation of Electron-Rich Aromatics with Sodium Arylsulfinates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Prevalence, Risk Factors, and Medical Costs of Chlamydia Trachomatis
Huai et al. BMC Infectious Diseases (2018) 18:534 https://doi.org/10.1186/s12879-018-3432-y RESEARCH ARTICLE Open Access Prevalence, risk factors, and medical costs of Chlamydia trachomatis infections in Shandong Province, China: a population- based, cross-sectional study Pengcheng Huai1,2,3 , Furong Li2, Zhen Li2, Lele Sun2,4,Xi’an Fu2,4, Qing Pan2,4, Gongqi Yu2,4, Zemin Chai2,4, Tongsheng Chu2, Zihao Mi2,4, Fangfang Bao2,4, Honglei Wang2,4, Bingni Zhou2,4, Chuan Wang2,4, Yonghu Sun2,4, Guiye Niu2,4, Yuan Zhang2,4, Fanghui Fu2,4, Xiaoqiao Lang2,4, Xiaoling Wang2,4, Hui Zhao2,4, Daina Liu2,4, Hong Liu2,4, Dianchang Liu2, Jian Liu2, Aiqiang Xu3,5 and Furen Zhang2,4* Abstract Background: A population-based study of Chlamydia trachomatis (CT) infections is essential in designing a specific control program; however, no large investigation of CT infections among the general population in mainland China has been conducted since 2000. We aimed to determine the prevalence, risk factors, and associated medical costs of CT among residents, 18–49 years of age, in Shandong, China. Methods: From May to August 2016, a multistage probability sampling survey involving 8074 individuals was distributed. Data were collected via face-to-face interviews, followed by self-administered questionnaire surveys. First-void urines were collected and tested for CT and Neisseria gonorrhoeae (NG) using nucleic acid amplification. Results: The weighted prevalence of CT infection was 2.3% (95% confidence interval [CI], 1.5–3.2) in females and 2.7% (1. 6–3.8) in males. Women, 30–34 years of age, had the highest prevalence of CT infections (3.5%, 2.6–4.4), while the highest prevalence of CT infections in males was in those 18–24 years of age (4.3%, 0.0–8.8). -

2019年第3季度在韩国注册的中国水产企业名单the List of Chinese
2019年第3季度在韩国注册的中国水产企业名单 The List of Chinese Registered Fishery Processing Establishments Export to Korea (Total 1347 , the third quarter of 2019,updated on 25 June, 2019) No. Est.No. 企业名称(中文) Est.Name 企业地址(中文) Est.Address 产品(Products) 北京市朝阳区崔各庄乡 The 23rd floor Sanyuan Property Jingmi Road 北京中洋环球金枪鱼有 1 1100/02010 Beijing Zhongyang Global Tuna Co.,Ltd 东辛店村京密路三元物 Dongxindian Village Cuigezhuang TownChaoyang Fishery Products 限公司 业院内23号楼 District Beijng 五洋海产(天津)有限 天津市塘沽区东江路 2 1200/02004 Ocean Products (Tian.Jin) Co., Ltd Dongjiang Road No.3849 Tanggu Tianjin Fishery Products 公司 3849号 欧盛实业(天津)有限 天津经济技术开发区渤 No.5, Bohai Road, Tianjin Economic & Technological 3 1200/02019 Ocean (Tianjin) Corporation Ltd. Fishery Products 公司 海路5号 Development Area, Tianjin 天津市颖明海湾食品有 天津市滨海新区中心渔 No.221 Yuehai RD., Binhai New Area Of The City 4 1200/02028 Tianjin Smart Gulf Foodstuffs Co.,Ltd. Fishery Products 限公司 港经济区悦海路221号 Center Fishing Port Economic Zone, Tianjin, China 天津市塘沽区海华水产 Tianjin Tanggu District Haihua Fishery Products Food 天津市塘沽区北塘镇水 No. 9, Shuichan Road, Beitang Town, Tanggu District, 5 1200/02048 Fishery Products 食品有限公司 Co., Ltd. 产路9号 Tianjin 天津百迅达进出口贸易 天津市津南区双桥河镇 South Dongnigu Village, Shuangqiaohe Town, Jinnan 6 1200/02063 Tianjin baixunda import and export trade Co., Ltd Fishery Products 有限公司 东泥沽村南 District, Tianjin, China 昌黎县筑鑫实业有限公 秦皇岛市昌黎县新开口 Economic Development Zone Xinkaikou Changli 7 1300/02228 Changli Zhuxin Enterprises Co., Ltd. Fishery Products 司 经济开发区 County Qinhuangdao 抚宁县渤远水产品有限 秦皇岛市抚宁县台营镇 Yegezhuang village taiying town funing county 8 1300/02229 Funing county boyuan aquatic products co.,ltd Fishery Products 公司 埜各庄村 Qinhuangdao city Hebei province 秦皇岛市江鑫水产冷冻 河北省秦皇岛北戴河新 Nandaihe Second District,Beidaihe New 9 1300/02236 Qinhuangdao Jiangxin Aquatic Food Products Co., Ltd. -

Cereal Series/Protein Series Jiangxi Cowin Food Co., Ltd. Huangjindui
产品总称 委托方名称(英) 申请地址(英) Huangjindui Industrial Park, Shanggao County, Yichun City, Jiangxi Province, Cereal Series/Protein Series Jiangxi Cowin Food Co., Ltd. China Folic acid/D-calcium Pantothenate/Thiamine Mononitrate/Thiamine East of Huangdian Village (West of Tongxingfengan), Kenli Town, Kenli County, Hydrochloride/Riboflavin/Beta Alanine/Pyridoxine Xinfa Pharmaceutical Co., Ltd. Dongying City, Shandong Province, 257500, China Hydrochloride/Sucralose/Dexpanthenol LMZ Herbal Toothpaste Liuzhou LMZ Co.,Ltd. No.282 Donghuan Road,Liuzhou City,Guangxi,China Flavor/Seasoning Hubei Handyware Food Biotech Co.,Ltd. 6 Dongdi Road, Xiantao City, Hubei Province, China SODIUM CARBOXYMETHYL CELLULOSE(CMC) ANQIU EAGLE CELLULOSE CO., LTD Xinbingmaying Village, Linghe Town, Anqiu City, Weifang City, Shandong Province No. 569, Yingerle Road, Economic Development Zone, Qingyun County, Dezhou, biscuit Shandong Yingerle Hwa Tai Food Industry Co., Ltd Shandong, China (Mainland) Maltose, Malt Extract, Dry Malt Extract, Barley Extract Guangzhou Heliyuan Foodstuff Co.,LTD Mache Village, Shitan Town, Zengcheng, Guangzhou,Guangdong,China No.3, Xinxing Road, Wuqing Development Area, Tianjin Hi-tech Industrial Park, Non-Dairy Whip Topping\PREMIX Rich Bakery Products(Tianjin)Co.,Ltd. Tianjin, China. Edible oils and fats / Filling of foods/Milk Beverages TIANJIN YOSHIYOSHI FOOD CO., LTD. No. 52 Bohai Road, TEDA, Tianjin, China Solid beverage/Milk tea mate(Non dairy creamer)/Flavored 2nd phase of Diqiuhuanpo, Economic Development Zone, Deqing County, Huzhou Zhejiang Qiyiniao Biological Technology Co., Ltd. concentrated beverage/ Fruit jam/Bubble jam City, Zhejiang Province, P.R. China Solid beverage/Flavored concentrated beverage/Concentrated juice/ Hangzhou Jiahe Food Co.,Ltd No.5 Yaojia Road Gouzhuang Liangzhu Street Yuhang District Hangzhou Fruit Jam Production of Hydrolyzed Vegetable Protein Powder/Caramel Color/Red Fermented Rice Powder/Monascus Red Color/Monascus Yellow Shandong Zhonghui Biotechnology Co., Ltd. -
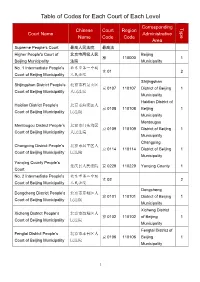
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

View Our Factory List
List updated 16th June 2021 FACTORY LIST Country Road Group is committed to driving positive social and environmental change in our supply chain. We mandate safe, inclusive and respectful workplaces wherever our products are manufactured, and are committed to greater transparency of our manufacturing operations. In line with this commitment, Country Road Group has publicly disclosed this list of factories involved in the manufacture of Country Road Group-branded products. This list includes the names, addresses, product categories and worker statistics of sites engaged in the manufacture of our goods. All sites listed are included in our Approved Factory Program and our team works closely with all suppliers and factories to continuously implement improvements. By releasing this information, we aim to provide customers with greater insights into our supply chain. All sites listed are correct at the time of publishing, and due to the seasonal nature of the retail industry this list is subject to change. * Data Not Captured: we are working to close this data gap through updated audit reports and working back with our supply partners. % OF % OF TOTAL SITE NAME SITE ADDRESS PROVINCE COUNTRY PRODUCT CATEGORY FEMALE INTERNATIONAL WORKERS WORKERS MIGRANT WORKERS ABHI HOME PLOT NO. 45, SECTOR- 29, PART – 1, HUDA, PANIPAT HARYANA INDIA HOME 129 8% * ABRAHAM MOON AND SONS LTD NETHERFIELD MILLS, GUISELEY, LEEDS WEST YORKSHIRE UNITED KINGDOM FOOTWEAR & ACCESSORIES, HOME 232 34% * ACME INDUSTRIES CO., LTD 99 MOO 4, BANGNA-TRAD KM. 35 BANGPLEE-NOI, BANGBOR SAMUT PRAKAN THAILAND HOME 436 55% 33% ANHUI TENGYANG CLOTHING CO., LTD. BUILDING 3#, YILITENG INDUSTRIAL PARK, QINGHE ROAD LUYANG INDUSTRIAL PARK, LUYANG DISTRICT, HEFEI ANHUI CHINA APPAREL 246 60% * ANM INTERNATIONAL PLOT NO.-62, SECTOR-6 IMT MANESAR, GURGAON HARYANA INDIA FOOTWEAR & ACCESSORIES 88 8% 0% ANTSIRABE KNITWEAR SA PROPRIETE VOION TN 1458 AMBOHIMENA, ANTSIRABE VAKINANKARATRA MADAGASCAR APPAREL 1653 43% * ART WAY CO., LTD. -

Everbright Greentech Secures Wendeng Biomass Electricity and Heat Cogeneration Project and Linshu Hazardous Waste Landfill Project
Press Release EVERBRIGHT GREENTECH SECURES WENDENG BIOMASS ELECTRICITY AND HEAT COGENERATION PROJECT AND LINSHU HAZARDOUS WASTE LANDFILL PROJECT Hong Kong, 26 June, 2017 - China Everbright Greentech Limited (“Everbright Greentech” or the “Company”) (HKSE: 01257) is pleased to announce that it has secured Wendeng Biomass Electricity and Heat Cogeneration Project (“Wendeng Project”) and Linshu Hazardous Waste Landfill Project (“Linshu Landfill Project”) in Shandong Province, the total investment of which will amount to approximately RMB479 million. According to the investment agreement signed by the Company and the People’s Government of Wendeng District in Weihai City of Shandong Province, Wendeng Project will be invested in and constructed on a BOO (Build-Operate-Own) model, and will command a total investment of approximately RMB330 million. The project is designed to have an annual biomass processing capacity of 300,000 tonnes, and is expected to generate around 180,000,000 kWh of green electricity annually. Meanwhile, the Company signed an investment agreement with the People’s Government of Linshu County in Shandong Province to invest in and construct Linshu Landfill Project in two phases. The project has a total designed storage capacity of 700,000 m3. Phase I of the project, with a total investment of approximately RMB149 million, will have a designed storage capacity of 300,000 m3 and a designed annual hazardous waste treatment capacity of 20,000 tonnes. Mr. Qian Xiaodong, CEO of Everbright Greentech, said, “Everbright Greentech has secured several new projects recently, which demonstrates our strong business development and efficient execution capabilities. Wendeng Project is the Company’s first biomass project in Shandong Province. -

Zhongrun Resources Investment Corporation Audit Report And
Zhongrun Resources Investment Corporation Audit Report and Financial Statements Financial Year 2019 Zhongrun Resources Investment Corporation Audit Report and Financial Statements (January 1, 2019 – December 31, 2019) Content Page 1. Audit report 1-6 2. Financial statements Consolidated Balance Sheet and Balance Sheet of Parent 1-6 company Consolidated Profit and Loss Statement and Profit and Loss 7-10 Statement of Parent Company Consolidated Cash Flow Statement and Cash Flow Statement of 11-14 Parent Company Consolidated Statement of Changes in Owner’s Equity and 15-23 Statement of Changes in Owner’s Equity of Parent Company Notes to Financial Statements 1-123 Audit Report BDO AR [2020] No. To all shareholders of Zhongrun Resources Investment Corporation: 1. Qualified Audit Opinion We have audited the attached financial statements of Zhongrun Resources Investment Corporation (ZRC), including the Consolidated Balance Sheet (dated 31 December 2019), Balance Sheet of Parent Company (dated 31 December 2019), Consolidated Profit and Loss Statement, Profit and Loss Statement of Parent Company, Consolidated Cash Flow Statement, Cash Flow Statement of Parent Company, Consolidated Statement of Changes in Owner’s Equity, Statement of Changes in Owner’s Equity of Parent Company, and Notes to Financial Statement. We believe that except the impact of the matters described in the “Basis for Formation of Qualified Audit Opinions” section, the attached financial statements were prepared in accordance with the requirements of the Accounting Standards for Business Enterprises in all material respects, and they have fairly reflected the financial positions of ZRC as of December 31, 2019 and the operating results and cash flow of ZRC in 2019, both on a consolidated basis and of the parent company. -

Original Article the Therapeutic Effects of the Minimally Invasive Repair of Hand Flexor Tendon Injuries
Int J Clin Exp Med 2019;12(12):13706-13711 www.ijcem.com /ISSN:1940-5901/IJCEM0101060 Original Article The therapeutic effects of the minimally invasive repair of hand flexor tendon injuries Jiqiang Cheng1, Yejin Feng2, Bin Liu1, Guiqing Gong1, Haiyong Yu3 1Third Department of Orthopaedics, 2Fourth Department of Orthopaedics, Ji’nan Zhangqiu District Hospital of Traditional Chinese Medicine, Ji’nan, Shandong Province, China; 3Department of Orthopedics, Wendeng Orthopaedic and Traumatologic Hospital of Shandong Province, Weihai, Shandong Province, China Received August 16 2019; Accepted October 10, 2019; Epub December 15, 2019; Published December 30, 2019 Abstract: Objective: To compare the therapeutic effects of the minimally invasive repair and conventional therapy of hand flexor tendon injuries. Methods: This study recruited 98 patients with flexor tendon injuries of the hand who were randomly divided into a control group (49 patients who received traditional surgery) and an experimental group (49 patients who were treated with minimally invasive repair). The excellent and good treatment rates, the complication rate, the visual simulation self-assessment scale (VAS) score, the patient satisfaction rate, and the short form 36 questionnaire (SF-36) scale were compared between the two groups. Results: The excellent and good treatment rates, the patient satisfaction rate, and the SF-36 scale in the experimental group were significantly higher than they were in the control group (all P<0.05). Meanwhile, the complication rate and the VAS score in the experimental group were significantly decreased compared with the control group (all P<0.05). Conclusion: For patients with flexor tendon injury, a minimally invasive repair can effectively improve hand function, reduce the incidence of complications, and raise the quality of life. -

List of Business Partners and Factories – October 2020
Otto Group – List of business partners and factories – October 2020 This list contains business partners (only private labels) as well as the final production factories, which have been active for the Otto Group companies bonprix, Otto, myToys, Heine, Schwab and/or Witt. A business partner/factory is considered active if it has been active within the past 12 months and remains active as of the date the list is created. Only factories that are located in so-called risk countries according to the amfori BSCI classification are included. The Otto Group also produces in non-risk countries, e.g. the EU. All factory related information is based on data that suppliers share with Otto Group companies. The list is updated regularly but not on a daily basis. Type of Supplier Name Country City Factory Address Type of Social Audit/Certificate Business Partner 3S IMPORT & EXPORT SHIJIA CO., LTD China Shijiazhuang n.a. n.a. Business Partner A&R MODEN GMBH Germany Loerrach n.a. n.a. Business Partner A.KUDRESOVO FIRMA Lithuania Kaunas n.a. n.a. Business Partner AANYA DESIGNS MANUFACTURERS & EXPORTERS India Moradabad n.a. n.a. Business Partner AB KAUNO BALDAI Lithuania Kaunas n.a. n.a. Business Partner ABG24 Spolka z ograniczona odpowiedzialnosic (0010053817) Poland Lodz n.a. n.a. Business Partner ACTONA COMPANY A/S Denmark Holstebro n.a. n.a. Business Partner ADALTEKS LTD Bulgaria Sofia n.a. n.a. Business Partner ADAM EXPORTS SYNTHOFINE IND. ESTATE, B (0020010395) India Mumbai n.a. n.a. Business Partner ADIYAMAN DENIZ TEKSTIL SAN VE DIS TIC. -

Primark Does Not Own Any Factories and Is Selective About the Suppliers with Whom We Work
Information last updated November 2020 Global Sourcing Map Factory List - Bangladesh Factory Name Address Country Total Men Women Number of Workers A&A Trousers Ltd Haribaritek Pubail College Gate Gazipur Dhaka 1721 Bangladesh 2250 996 (44.3)% 1254 (55.7)% Afiya Knitwear Ltd 10/ 2 Durgapur Ashulia Savar Dhaka 1341 Bangladesh 496 232 (46.8)% 264 (53.2)% AKH Apparels Ltd 128 Hemayetpur Savar Dhaka 1340 Bangladesh 2136 855 (40.0)% 1281 (60.0)% Alpha Clothing Ltd Tenguri, BKSP Ashulia Savar Dhaka Bangladesh 1971 968 (49.1)% 1003 (50.9)% Ananta Huaxiang Ltd 222, 223, H2-H4, Adamjee EPZ Shiddirgonj Narayanganj Dhaka Bangladesh 2038 982 (48.2)% 1056 (51.8)% Anowara Knit Composite Ltd Mulaid Mawna Sreepur Gazipur Bangladesh 2276 1329 (58.4)% 947 (41.6)% Aspire Garments Ltd 491 Dhalla Singair Manikganj 1822 Bangladesh 2992 1389 (46.4)% 1603 (53.6)% ASR Sweater Limited Mulaied Maona Sreepur Gazipur Bangladesh 1458 927 (63.6)% 531 (36.4)% Primark does not own any factories and is selective about the suppliers with whom we work. Every factory which manufactures product for Primark has to commit to meeting internationally recognised standards, before the first order is placed and throughout the time they work with us. The factories featured on Primark’s Global Sourcing Map are Primark’s suppliers’ production sites which represent over 95% of Primark products for sale in Primark stores. A factory is detailed on the Map only after it has produced products for Primark for a year and has become an established supplier. During the first year a factory has to demonstrate that it can consistently work to Primark’s ethical standards, as well as meet our commercial requirements in areas such as quality and timely delivery. -

Producent Adres Land
*Deze lijst bevat alle 'non-food' leveranciers die producten aan Lidl hebben geleverd in de periode tussen 1 maart 2019 en 29 februari 2020. Producent Adres Land 3W Home Fashion Heyuan Co., Ltd. Mingzhu Industrial Park, Heyuan West Road, Chuangye South Road, Heyuan, Guangdong China A.B Sales Corp. (A Unit Of Satyam Creations (Pvt) Ltd.) Plot No. 1642, Zone -09, Kolkata Leather Complex, Bantala, 24 Parganas (South), Kolkatta, West Bengal India AB Apparels Ltd. No. 225 Singair Road, Tetuljhora, Hemayetpur, Savar, Dhaka Bangladesh Above & Beyond Co., Ltd. Plot No. 116/A, 116/B, Settmu (10) Street, Myay Taing Block No. 42, Industrial Zone (1), Shwe Pyi Thar Township, Yangon Myanmar Ador Composite Ltd. 1, C&B Bazar, Gilarchala, Sreepur, Gazipur, Bd Gazipur District, Gazipur, Dhaka Bangladesh Advanced Composite Textile Ltd. Kashor Masterbari, Bhaluka, Mymensingh, Sylhet Bangladesh Afroze Textile Industries (Pvt) Ltd. Plot C-8, Scheme 33, S.I.T.E., Super Highway, Karachi, Sindh Pakistan Afroze Textile Industries (Pvt) Ltd. LA-1/A, Block 22, F.B. Area, Karachi, Sindh Pakistan Ahmed Fashions 34/1, Darus Salam Road, Dhaka Bangladesh Ai Qi Fujian Shoes Plastic Co., Ltd. Meiling Street, Shuanggou Industrial District, Sichuan, Jinjiang, Fujian China Al Hadi Textile (Pvt) Ltd. D-12 Site Super Highway Industrial Area, Karachi, Sindh Pakistan Alpine Clothings Polpithigama (Pvt) Ltd. Andarayaya, Polpithigama Sri Lanka Alpine Clothings Yapahuwa (Pvt) Ltd. Anuradhapura Road, Uduweriya, North Western Sri Lanka AMG Factory Ltd. Plot No. 51 & 52, Myay Taing Block No. 25, Shwe Lin Ban Industrial Zone, Hlaing Thar Yar Township, Yangon Myanmar Andy Accessory Co., Ltd. -

Global Sourcing Map Factory List - Bangladesh
Information last updated July 2018 Global Sourcing Map Factory List - Bangladesh Factory Name Address Country Total Men Women Number of Workers Afiya Knitwear Ltd 10/ 2 Durgapur Ashulia Savar Dhaka 1341 Bangladesh 579 288 (49.7)% 291 (50.3)% AKH Apparels Ltd 128 Hemayetpur Savar Dhaka 1340 Bangladesh 2188 971 (44.4)% 1217 (55.6)% AKH Eco Apparels Ltd 495 Balitha Shah-Belishwer Dhamrai Dhaka 1800 Bangladesh 5418 3000 (55.4)% 2418 (44.6)% Alpha Knitting Wear Limited 888 Shewrapara, 1St & 2Nd Floor Begum Rokeya Shoroni Mirpur Dhaka 1216 Bangladesh 2307 682 (29.6)% 1625 (70.4)% Ananta Denim Technology Ltd Noyabari Kanchpur Sonargaon Narayanganj Bangladesh 4833 2394 (49.5)% 2439 (50.5)% Ananta Huaxiang Ltd 222, 223, H2-H4, Adamjee Epz Shiddirgonj Narayanganj Dhaka Bangladesh 2039 1184 (58.1)% 855 (41.9)% Anowara Knit Composite Ltd Mulaid Mawna Sreepur Gazipur Bangladesh 1979 1088 (55.0)% 891 (45.0)% Apex Lingerie Limited Chandora Kaliakoir Gazipur Bangladesh 3937 2041 (51.8)% 1896 (48.2)% Arunima Sportswear Ltd Dewan Idris Road Zirabo Ashulia Savar Dhaka Bangladesh 4481 2561 (57.2)% 1920 (42.8)% Primark does not own any factories and is selective about the suppliers with whom we work. Every factory which manufactures product for Primark has to commit to meeting internationally recognised standards, before the first order is placed and throughout the time they work with us. The factories featured on Primark’s Global Sourcing Map are Primark’s suppliers’ production sites which represent over 95% of Primark products for sale in Primark stores. A factory is detailed on the Map only after it has produced products for Primark for a year and has become an established supplier.