Nickel-Catalyzed Anti-Markovnikov Hydroarylation of Unactivated Alkenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Including Toluene Derivatives
RSC Advances View Article Online PAPER View Journal | View Issue Catalytic C–H aerobic and oxidant-induced oxidation of alkylbenzenes (including toluene Cite this: RSC Adv., 2020, 10,23543 derivatives) over VO2+ immobilized on core–shell Fe3O4@SiO2 at room temperature in water† Pegah Mohammadpour and Elham Safaei * Direct C–H bond oxidation of organic materials, and producing the necessary oxygenated compounds under mild conditions, has attracted increasing interest. The selective oxidation of various alkylbenzenes 2+ was carried out by means of a new catalyst containing VO species supported on silica-coated Fe3O4 nanoparticles using t-butyl hydroperoxide as an oxidant at room temperature in H2O or solvent-free media. The chemical and structural characterization of the catalyst using several methods such as FTIR spectroscopy, XRD, FETEM, FESEM, SAED, EDX and XPS showed that VO2+ is covalently bonded to the silica surface. High selectivity and excellent conversion of various toluene derivatives, with less reactive Creative Commons Attribution 3.0 Unported Licence. Received 18th April 2020 aliphatic (sp3)C–H bonds, to related benzoic acids were quite noticeable. The aerobic oxygenation Accepted 15th May 2020 reaction of these alkylbenzenes was studied under the same conditions. All the results accompanied by DOI: 10.1039/d0ra03483e sustainability of the inexpensive and simple magnetically separable heterogeneous catalyst proved the rsc.li/rsc-advances important criteria for commercial applications. Introduction industrial communities. Among -

Reactions of Benzene Or Alkylbenzenes with Steam Over a Silica-Supported Nickel Catalyst*
54 Reactions of Benzene or Alkylbenzenes with Steam over a Silica-supported Nickel Catalyst* by .Masahiro Saito**, Yoshio Sohda** Michiaki Tokuno** and Yoshiro Morita** Summary: Benzene-steam and alkylbenzenes-steam reactions over a silica-supported nickel catalyst have been studied under atmospheric pressure in a temperature range of 370~430℃. In the reactionof benzene-steam,the methaneyield is lower and the carbon dioxideyield is higher than the estimatedvalue. The reactionis zero order with respectto benzeneand approxi- mately first order with respect to steam. In the reactionof alkylbenzenes-steam,ring breakdownand dealkylationoccur at the initial stage of reaction, and theformer occursmore easily than the latter at high conversionand at high temperature. The number and the position of the alkyl group on the benzene ring influencethe reaction rate and the selectivity. The reactionpath of dealkylationis proposed as follows: most effective catalyst for dealkylation with 1 Introduction steam, and they investigated reactivity and reac- The catalytic reaction of hydrocarbon with tion path on Ni/BeO at 450℃. steam is important for the production of hydrogen, In the present work, the reactions of benzene synthesis gas and town gas. The authors have or alkylbenzenes with steam were carried out previously reported the works on olefins-steam over a silica-supported nickel catalyst under and paraffins-steam reactions over a silica-sup- atmospheric pressure in a temperature range of ported nickel catalyst1)~3), and this study on 370~430℃, and the effects of reaction conditions, aromatics-steam reactions has been carried out rate of each reaction and the reaction path were as a consecutive one. -

Disproportionation and Transalkylation of Alkylbenzenes Over Zeolite Catalysts
Applied Catalysis A: General 181 (1999) 355±398 Disproportionation and transalkylation of alkylbenzenes over zeolite catalysts Tseng-Chang Tsaia, Shang-Bin Liub, Ikai Wangc,* aRe®ning and Manufacturing Research Center, Chinese Petroleum Corporation, Chiayi 600, Taiwan bInstitute of Atomic and Molecular Sciences, Academia Sinica, PO Box 23-166, Taipei 106, Taiwan cDepartment of Chemical Engineering, National Tsing-Hua University, Hsinchu 300, Taiwan Received 13 June 1998; received in revised form 3 October 1998; accepted 5 November 1998 Abstract Disproportionation and transalkylation are important processes for the interconversion of mono-, di-, and tri-alkylbenzenes. In this review, we discuss the recent advances in process technology with special focus on improvements of para-isomer selectivity and catalyst stability. Extensive patent search and discussion on technology development are presented. The key criteria for process development are identi®ed. The working principles of para-isomer selectivity improvements involve the reduction of diffusivity and the inactivation of external surface. In conjunction with the fundamental research, various practical modi®cation aspects particularly the pre-coking and the silica deposition techniques, are extensively reviewed. The impact of para-isomer selective technology on process economics and product recovery strategy is discussed. Furthermore, perspective trends in related research and development are provided. # 1999 Elsevier Science B.V. All rights reserved. Keywords: Disproportionation; Transalkylation; -

Trimethylbenzenes CAS Registry Numbers: 526-73-6 (1,2,3-TMB) 95-63-6 (1,2,4-TMB) 108-67-8 (1,3,5-TMB) 25551-13-7 (Mixed Isomers)
Development Support Document Final, September 4, 2015 Trimethylbenzenes CAS Registry Numbers: 526-73-6 (1,2,3-TMB) 95-63-6 (1,2,4-TMB) 108-67-8 (1,3,5-TMB) 25551-13-7 (Mixed Isomers) Prepared by Joseph T. Haney, Jr., M.S. Angela Curry, M.S. Toxicology Division Office of the Executive Director TEXAS COMMISSION ON ENVIRONMENTAL QUALITY Trimethylbenzenes Page i TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................................................................ I LIST OF TABLES ......................................................................................................................................................II ACRONYMS AND ABBREVIATIONS ................................................................................................................. III CHAPTER 1 SUMMARY TABLES .......................................................................................................................... 1 CHAPTER 2 MAJOR SOURCES AND USES ......................................................................................................... 4 CHAPTER 3 ACUTE EVALUATION ...................................................................................................................... 4 ACUTE 3.1 HEALTH-BASED ACUTE REV AND ESL ........................................................................................................ 4 3.1.1 Physical/Chemical Properties .................................................................................................................... -

Studies of the Temporary Anion States of Unsaturated Hydrocarbons by Electron Transmission Spectroscopy
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Paul Burrow Publications Research Papers in Physics and Astronomy 1978 Studies of the Temporary Anion States of Unsaturated Hydrocarbons by Electron Transmission Spectroscopy Kenneth D. Jordan Paul Burrow Follow this and additional works at: https://digitalcommons.unl.edu/physicsburrow Part of the Atomic, Molecular and Optical Physics Commons This Article is brought to you for free and open access by the Research Papers in Physics and Astronomy at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Paul Burrow Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. digitalcommons.unl.edu Studies of the Temporary Anion States of Unsaturated Hydrocarbons by Electron Transmission Spectroscopy Kenneth D. Jordan Mason Laboratory, Department of Engineering and Applied Science, Yale University, New Haven, Connecticut 06520 Paul D. Burrow Behlen Laboratory of Physics, University of Nebraska, Lincoln, Nebraska 68588 The concept of occupied and unoccupied orbitals has provided a useful means for visualizing many of the most important properties of mo- lecular systems. Yet, there is a curious imbalance in our experimen- tal knowledge of the energies of occupied and unoccupied orbitals. Whereas photoelectron spectroscopy has provided a wealth of data on positive ion states and has established that they can be associated, within the context of Koopmans’ theorem, with the occupied orbitals of the neutral molecule, the corresponding information for the neg- ative ion states, associated with the normally unoccupied orbitals, is sparse. In part this reflects the experimental difficulties connected with measuring the electron affinities of molecules which possess sta- ble anions. -

BENZENE Disclaimer
United States Office of Air Quality EPA-454/R-98-011 Environmental Protection Planning And Standards June 1998 Agency Research Triangle Park, NC 27711 AIR EPA LOCATING AND ESTIMATING AIR EMISSIONS FROM SOURCES OF BENZENE Disclaimer This report has been reviewed by the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, and has been approved for publication. Mention of trade names and commercial products does not constitute endorsement or recommendation of use. EPA-454/R-98-011 ii TABLE OF CONTENTS Section Page LIST OF TABLES.....................................................x LIST OF FIGURES.................................................. xvi EXECUTIVE SUMMARY.............................................xx 1.0 PURPOSE OF DOCUMENT .......................................... 1-1 2.0 OVERVIEW OF DOCUMENT CONTENTS.............................. 2-1 3.0 BACKGROUND INFORMATION ...................................... 3-1 3.1 NATURE OF POLLUTANT..................................... 3-1 3.2 OVERVIEW OF PRODUCTION AND USE ......................... 3-4 3.3 OVERVIEW OF EMISSIONS.................................... 3-8 4.0 EMISSIONS FROM BENZENE PRODUCTION ........................... 4-1 4.1 CATALYTIC REFORMING/SEPARATION PROCESS................ 4-7 4.1.1 Process Description for Catalytic Reforming/Separation........... 4-7 4.1.2 Benzene Emissions from Catalytic Reforming/Separation .......... 4-9 4.2 TOLUENE DEALKYLATION AND TOLUENE DISPROPORTIONATION PROCESS ............................ 4-11 4.2.1 Toluene Dealkylation -

Classification Tests for Hydrocarbons Using Solubility, Ignition, Nitration, Baeyer’S Test, Bromine Test and Basic Oxidation Test
CLASSIFICATION TESTS FOR HYDROCARBONS USING SOLUBILITY, IGNITION, NITRATION, BAEYER’S TEST, BROMINE TEST AND BASIC OXIDATION TEST Jasher Christian Boado, Alyanna Cacas, Phoebe Calimag, Caryl Angelica Chin, Haidee Cosilet, John Francis Creencia Group 2, 2BMT, Faculty of Pharmacy, University of Santo Tomas ABSTRACT Hydrocarbons are classified as saturated, actively unsaturated, aromatic or an arene based on various classification tests involving test for solubility in concentrated H2SO4, ignition, active unsaturation using Baeyer’s test and Bromine test, aromaticity using nitration test, and basic oxidation test. This experiment aims to differentiate the intrinsic physical and the chemical properties of hydrocarbons, and to determine if it is saturated, actively unsaturated, aromatic or an arene. The sample compounds hexane, heptane, cyclohexane, cyclohexene, benzene and toluene were analyzed for their physical state in room temperature, color, and odor. Using solubility test, a drop of a sample was added cautiously into 1ml of concentrated H2SO4. Using the Baeyer’s Test and/or Bromine test in which 2 drops of 2% KmnO4 solution and 10 drops of 0.5% Br2 in CCl4 reagent, respectively, was added into 5 drops of a sample in a dry test tube, was shaken vigourously until the reagent is decolorized compared with water. Using Nitration test, 8 drops of nitrating mixture was added into 5 drops of a sample in a dry test tube, was observed for the formation of a yellow oily layer or droplet and was diluted with 20 drops of water. Using Basic Oxidation test, 8 drops of 2% KmnO4 solution and 3 drops of 10% NaOH solution was added into 4 drops of a sample in a dry test tube and was heated in a water bath for 2 minutes. -

And Nitro- Oxy-Polycyclic Aromatic Hydrocarbons
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization or the World Health Organization. Environmental Health Criteria 229 SELECTED NITRO- AND NITRO- OXY-POLYCYCLIC AROMATIC HYDROCARBONS First draft prepared by Drs J. Kielhorn, U. Wahnschaffe and I. Mangelsdorf, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany Please note that the pagination and layout of this web version are not identical to the printed EHC Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO) and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research and the Organisation for Economic Co- operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

Disproportionation of 1,2,4-Trimethylbenzene Over Zeolite NU-87
Korean J. Chem. Eng., 17(2), 198-204 (2000) Disproportionation of 1,2,4-Trimethylbenzene over Zeolite NU-87 Se-Ho Park, Jong-Hyung Lee and Hyun-Ku Rhee† School of Chemical Engineering and Institute of Chemical Processes, Seoul National University, Kwanak-ku, Seoul 151-742, Korea (Received 27 September 1999 • accepted 30 December 1999) Abstract−The catalytic properties of zeolite NU-87 were investigated with respect to the disproportionation of 1, 2,4-trimethylbenzene and the results were compared to those obtained over H-beta and H-mordenite with 12-mem- bered ring channel system. In the conversion of 1,2,4-trimethylbenzene, the disproportionation to xylene and tetra- methylbenzene is preferred to the isomerization into 1,2,3- and 1,3,5-isomers over all the three zeolites, but this trend is much more pronounced over HNU-87 owing to its peculiar pore structure. Disproportionation reaction is found to proceed within the micropores of HNU-87, whereas isomerization occurs mainly on the external surface. The high selectivity to disproportionation gives more xylenes and tetramethylbenzenes over HNU-87. The detailed descrip- tions for the product distribution are also reported. Key words: NU-87, 1,2,4-Trimethylbenzene, Disproportionation, Isomerization INTRODUCTION EXPERIMENTAL Disproportionation of trimethylbenzene (TMB) to xylene and 1. Catalysts Preparation = tetramethylbenzene (TeMB) is an important process for the indus- H-mordenite (Engelhard, SiO2/Al2O3 45) and H-beta (PQ Corp., = try, mainly due to the increasing demand for p-xylene to be used SiO2/Al2O3 22) used in this study were taken from commercial for the production of polyester resins. -

Industrial Hydrocarbon Processes
Handbook of INDUSTRIAL HYDROCARBON PROCESSES JAMES G. SPEIGHT PhD, DSc AMSTERDAM • BOSTON • HEIDELBERG • LONDON NEW YORK • OXFORD • PARIS • SAN DIEGO SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO Gulf Professional Publishing is an imprint of Elsevier Gulf Professional Publishing is an imprint of Elsevier The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, UK 30 Corporate Drive, Suite 400, Burlington, MA 01803, USA First edition 2011 Copyright Ó 2011 Elsevier Inc. All rights reserved No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the publisher Permissions may be sought directly from Elsevier’s Science & Technology Rights Department in Oxford, UK: phone (+44) (0) 1865 843830; fax (+44) (0) 1865 853333; email: [email protected]. Alternatively you can submit your request online by visiting the Elsevier web site at http://elsevier.com/locate/ permissions, and selecting Obtaining permission to use Elsevier material Notice No responsibility is assumed by the publisher for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any methods, products, instructions or ideas contained in the material herein. Because of rapid advances in the medical sciences, in particular, independent verification of diagnoses and drug dosages should be made British Library Cataloguing in Publication Data -
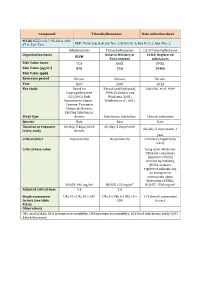
Compound Trimethylbenzenes Data Collection Sheet N°CAS
Compound Trimethylbenzenes Data collection sheet N°CAS 25551-13-7, 95-63-6, 108- 67-8, 526-73-8 CLP: Flam. Liq. 3, Acute Tox. 4, Skin Irrit. 2, Eye Irrit. 2, Asp. Tox. 1, Alkylbenzenes Trimethylbenzenes 1,2,4-Trimethylbenzene Organization Name Ontario Ministry of ECHA: Registered RIVM Environment substances Risk Value Name TCA AAQC DNEL Risk Value (µg/m3) 870 220 29400 Risk Value (ppb) Reference period Chronic Chronic Chronic Year 2007 2007 2013 Key Study Based on Korsak and Rydzynski, Clark DG, et al. 1989 Isopropylbenzene 1996; Gralewicz and EU (2001) Risk Wiaderna, 2001; Assessement Report – Wiaderna et al., 2002 Cumene. European Chemicals Bureau, Existing Substances. Study type chronic Subchronic inhalation Chronic inhalation Species Rats Rats Rats Duration of exposure 6h/day, 5 days/week 6h/day, 5 days/week 6h/day, 5 days/week, 1 in key study chronic year Critical effect Neurotoxicity Neurotoxicity Irritation (respiratory tract) Critical dose value Long term inhalation DNEL for consumers (systemic effects) derived by industry (ECHA-website: registered substances), no transparent information about derivation of DNEL NOAEL 490 mg/m3 NOAEL 123 mg/m³ NOAEC 1800 mg/m3 Adjusted critical dose 5.6 5.6 Single assessment UFH 10 x UFA 10 = 100 UFs 3 x UFH 3 x UFA 10 = 1.7 (Overall assessment factors (see table 100 factor) R.8.6) Other effects UFL used LOAEL; UFH intraspecies variability; UFA interspecies variability; UFS Used subchronic study; UFD data deficiencies Compound TRIMETHYLBENZENES Factsheet Parameter Note Comments Value / descriptor EU-LCI Value and Status EU-LCI value 1 Mass/volume [µg/m³] 450 EU-LCI status 2 Draft / Final Final Year when the EU-LCI value has been EU-LCI year of issue 3 2012 issued General Information 601-025-00-5 CLP-INDEX-Nr. -

Diesel Oil #1 Environmental Hazard Summary
ENVIRONMENTAL CONTAMINANTS ENCYCLOPEDIA DIESEL OIL #1 ENTRY July 1, 1997 COMPILERS/EDITORS: ROY J. IRWIN, NATIONAL PARK SERVICE WITH ASSISTANCE FROM COLORADO STATE UNIVERSITY STUDENT ASSISTANT CONTAMINANTS SPECIALISTS: MARK VAN MOUWERIK LYNETTE STEVENS MARION DUBLER SEESE WENDY BASHAM NATIONAL PARK SERVICE WATER RESOURCES DIVISIONS, WATER OPERATIONS BRANCH 1201 Oakridge Drive, Suite 250 FORT COLLINS, COLORADO 80525 WARNING/DISCLAIMERS: Where specific products, books, or laboratories are mentioned, no official U.S. government endorsement is implied. Digital format users: No software was independently developed for this project. Technical questions related to software should be directed to the manufacturer of whatever software is being used to read the files. Adobe Acrobat PDF files are supplied to allow use of this product with a wide variety of software and hardware (DOS, Windows, MAC, and UNIX). This document was put together by human beings, mostly by compiling or summarizing what other human beings have written. Therefore, it most likely contains some mistakes and/or potential misinterpretations and should be used primarily as a way to search quickly for basic information and information sources. It should not be viewed as an exhaustive, "last-word" source for critical applications (such as those requiring legally defensible information). For critical applications (such as litigation applications), it is best to use this document to find sources, and then to obtain the original documents and/or talk to the authors before depending too heavily on a particular piece of information. Like a library or most large databases (such as EPA's national STORET water quality database), this document contains information of variable quality from very diverse sources.