The Role of Bile Acids in Overcoming Resistance to Chemotherapy By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Farnesyl Phosphatase, a Corpora Allata
Florida International University FIU Digital Commons Department of Biological Sciences College of Arts, Sciences & Education 8-5-2013 Farnesyl Phosphatase, a Corpora allata Enzyme Involved in Juvenile Hormone Biosynthesis in Aedes aegypti Pratik Nyati Department of Biological Sciences, Florida International University, [email protected] Marcela Nouzova Department of Biological Sciences, Florida International University, [email protected] Crisalejandra Rivera-Perez Department of Biological Sciences, Florida International University,, [email protected] Mark E. Clifton Department of Biological Sciences, Florida International University Jamie G. Mayoral Department of Biological Sciences, Florida International University See next page for additional authors Follow this and additional works at: https://digitalcommons.fiu.edu/cas_bio Part of the Biology Commons Recommended Citation Nyati P, Nouzova M, Rivera-Perez C, Clifton ME, Mayoral JG, et al. (2013) Farnesyl Phosphatase, a Corpora allata Enzyme Involved in Juvenile Hormone Biosynthesis in Aedes aegypti. PLoS ONE 8(8): e71967. doi:10.1371/journal.pone.0071967 This work is brought to you for free and open access by the College of Arts, Sciences & Education at FIU Digital Commons. It has been accepted for inclusion in Department of Biological Sciences by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. Authors Pratik Nyati, Marcela Nouzova, Crisalejandra Rivera-Perez, Mark E. Clifton, Jamie G. Mayoral, and Fernando G. Noriega This article is available at FIU Digital Commons: https://digitalcommons.fiu.edu/cas_bio/131 Farnesyl Phosphatase, a Corpora allata Enzyme Involved in Juvenile Hormone Biosynthesis in Aedes aegypti Pratik Nyati, Marcela Nouzova, Crisalejandra Rivera-Perez, Mark E. Clifton, Jaime G. Mayoral, Fernando G. -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

Characterization of Juvenile Hormone Biosynthetic Enzymes in the Mosquito, Aedes Aegypti Pratik Nyati Florida International University, [email protected]
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-5-2014 Characterization of Juvenile Hormone Biosynthetic Enzymes in the Mosquito, Aedes aegypti Pratik Nyati Florida International University, [email protected] Follow this and additional works at: http://digitalcommons.fiu.edu/etd Part of the Biochemistry, Biophysics, and Structural Biology Commons, Biology Commons, and the Entomology Commons Recommended Citation Nyati, Pratik, "Characterization of Juvenile Hormone Biosynthetic Enzymes in the Mosquito, Aedes aegypti" (2014). FIU Electronic Theses and Dissertations. Paper 1688. http://digitalcommons.fiu.edu/etd/1688 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida CHARACTERIZATION OF JUVENILE HORMONE BIOSYNTHETIC ENZYMES IN THE MOSQUITO, AEDES AEGYPTI A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in BIOLOGY by Pratik Nyati 2014 To: Interim Dean Michael R. Heithaus College of Arts and Sciences This dissertation, written by Pratik Nyati, and entitled Characterization of Juvenile Hormone Biosynthetic Enzymes in the Mosquito, Aedes aegypti, having been approved in respect to style and intellectual content, is referred to you for judgment. We have read this dissertation and recommend that it be approved. ______________________________________ Alejandro Barbieri ______________________________________ Lidia Kos ______________________________________ Lou W. Kim ______________________________________ Kalai Mathee ______________________________________ Fernando G. Noriega, Major Professor Date of Defense: November 5, 2014 This dissertation of Pratik Nyati is approved. -

9, 10-Phenanthrenequinone Induces Monocytic Differentiation of U937
1598 Note Biol. Pharm. Bull. 35(9) 1598–1602 (2012) Vol. 35, No. 9 9,10-Phenanthrenequinone Induces Monocytic Differentiation of U937 Cells through Regulating Expression of Aldo-Keto Reductase 1C3 Toshiyuki Matsunaga,*,a Mika Hosogai,a Marina Arakaki,a Satoshi Endo,a Ossama El-Kabbani,b and Akira Haraa a Laboratory of Biochemistry, Gifu Pharmaceutical University; Gifu 501–1196, Japan: and b Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences; Victoria 3052, Australia. Received March 9, 2012; accepted June 5, 2012 Persistent inhalation of diesel exhaust particles results in damaged lung cells through formation of re- active oxygen species (ROS), but the details of the toxicity mechanism against monocytes are poorly under- stood. In this study, we used human promyelomonocytic U937 cells as surrogates of monocytes and investi- gated the toxicity mechanism initiated by exposure to 9,10-phenanthrenequinone (9,10-PQ), a major quinone component in diesel exhaust particles. A 24-h incubation with 9,10-PQ provoked apoptotic cell death, which was due to signaling through the enhanced ROS generation and concomitant caspase activation. Flow cy- tometric analyses of U937 cells after long-term exposure to 9,10-PQ revealed induction of differentiation that was evidenced by increasing expression of CD11b/CD18, a cell-surface marker for monocytic differentiation into macrophages. The 9,10-PQ-induced differentiation was significantly abolished by ROS inhibitors, sug- gesting that ROS generation contributes to cell differentiation. The 9,10-PQ treatment increased the expres- sion of aldo-keto reductase (AKR) 1C3, which reached a peak at 1 to 2 d post-treatment and then declined. -

Ep 2679685 A1
(19) TZZ _T (11) EP 2 679 685 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 01.01.2014 Bulletin 2014/01 C12N 15/52 (2006.01) C12P 7/18 (2006.01) C12N 1/00 (2006.01) (21) Application number: 13001196.8 (22) Date of filing: 06.08.2008 (84) Designated Contracting States: • Van Dien, Stephen J. AT BE BG CH CY CZ DE DK EE ES FI FR GB GR San Diego, CA 92121 (US) HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT •Burk,Mark J. RO SE SI SK TR San Diego, CA 92121 (US) (30) Priority: 10.08.2007 US 891602 (74) Representative: HOFFMANN EITLE Patent- und Rechtsanwälte (62) Document number(s) of the earlier application(s) in Arabellastrasse 4 accordance with Art. 76 EPC: 81925 München (DE) 08782639.2 / 2 185 708 Remarks: (71) Applicant: Genomatica, Inc. •This application was filed on 11-03-2013 as a San Diego, CA 92121 (US) divisional application to the application mentioned under INID code 62. (72) Inventors: •Claims filed after the date of filing of the application/ • Burgard, Anthony P. after the date of receipt of the divisional application San Diego, CA 92121 (US) (Rule 68(4) EPC). (54) Methods and organisms for the growth- coupled production of 1,4-butanediol (57) The invention provides a non- naturally occurring microorganismcomprising one ormore gene disruptions, the one or more gene disruptions occurring in genes en- coding an enzyme obligatory to coupling 1,4-butanediol production to growth of the microorganism when the gene disruption reduces an activity of the enzyme, whereby theone or more gene disruptions confers stable growth- coupled production of 1,4- butanediol onto the non-natu- rally occurring microorganism. -

Investigating Labdane-Related Diterpenoids Biosynthetic Network In
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2012 Investigating labdane-related diterpenoids biosynthetic network in rice: characterization of cytochromes P450 and short-chain alcohol dehydrogenases/reductase Yisheng Wu Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Biochemistry Commons Recommended Citation Wu, Yisheng, "Investigating labdane-related diterpenoids biosynthetic network in rice: characterization of cytochromes P450 and short-chain alcohol dehydrogenases/reductase" (2012). Graduate Theses and Dissertations. 12680. https://lib.dr.iastate.edu/etd/12680 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Investigating labdane-related diterpenoids biosynthetic network in rice: Characterization of cytochromes P450 and short-chain alcohol dehydrogenases/reductase by Yisheng Wu A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Biochemistry Program of Study Committee: Reuben Peters, Major Professor Gustavo MacIntosh Thomas Bobik Bing Yang Mark Hargrove Iowa State University Ames, Iowa 2012 Copyright © Yisheng Wu , 2012. All -
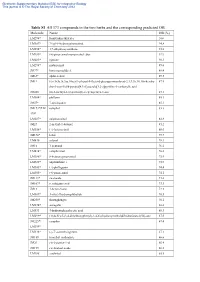
Table S1 All 373 Compounds in the Two Herbs and the Corresponding
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Table S1 All 373 compounds in the two herbs and the corresponding predicted OB Molecule Name OB (%) LM294* forsythidmethylester 100 LM387* 7'-epi-8-hydroxypinoresinol 94.8 LM394* 1,7-dihydroxyxanthone 93.8 LM289* (+)-pinoresinol monomethyl ether 92.9 LM285* egenine 90.3 LM298* matairesinol 89.6 JM77* loniceracetalide A 89.4 JM63* alpha-cedrol 89.3 JM1* (-)-(3r,8s,9r,9as,10as)-9-ethenyl-8-(beta-d-glucopyranosyloxy)-2,3,9,9a,10,10a-hexahy 87.5 dro-5-oxo-5h,8h-pyrano[4,3-d]oxazolo[3,2-a]pyridine-3-carboxylic acid JM200 (z)-3-methyl-2-(2-pentenyl)-2-cyclopenten-1-one 87.1 LM304* phillyrin 85.1 JM57* 7-epi-loganin 85.1 JM173*/LM camphol 83.3 398* LM287* epipinoresinol 82.8 JM27 2-methyl-1-butanol 81.2 LM356* (+)-lariciresinol 80.0 JM170* ledol 79.7 LM430 safynol 78.3 JM16 1-pentanol 76.2 LM414* campherenol 76.0 LM386* 8-hydroxypinoresinol 75.9 LM428* onjixanthone i 75.9 LM345* (+)-phillygenin 74.4 LM305* (+)-pinoresinol 74.1 JM113* sweroside 73.6 JM107* secologanic acid 73.3 JM13 1-hexen-3-one 72.3 LM309* 3-ethyl-7hydroxyphthalide 70.5 JM250* shuangkangsu 70.1 LM274* astragalin 68.6 LM331 4-hydroxyphenylacetic acid 68.3 LM299* (3r,4s,5r)-5-(3,4-dimethoxyphenyl)-3,4-bis(hydroxymethyl)dihydrofuran-2(3h)-one 67.5 JM225*/ camphor 67.4 LM399* LM338* (-)-7’-o-methylegenine 67.1 JM189 trimethyl isothiazole 66.6 JM29 cis-2-penten-1-ol 66.4 JM199 cis-linalool oxide 66.2 LM288 erythritol 65.8 Electronic Supplementary Material (ESI) for -

Functional and Structural Studies of AKR1B15 and AKR1B16: Two Novel Additions to Human and Mouse Aldo-Keto Reductase Superfamily
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Functional and structural studies of AKR1B15 and AKR1B16: Two novel additions to human and mouse aldo-keto reductase superfamily Mem`oriapresentada per JOAN GIMENEZ´ DEJOZ per optar al Grau de Doctor en Bioqu´ımicai Biologia Molecular Treball realitzat al departament de Bioqu´ımicai Biologia Molecular de la Universitat Aut`onomade Barcelona, sota la direcci´odels Doctors SERGIO PORTE´ ORDUNA, JAUME FARRES´ VICEN´ i XAVIER PARES´ CASASAMPERA Sergio Port´eOrduna Jaume Farr´esVic´en Xavier Par´esCasasampera Joan Gim´enezDejoz Bellaterra, 17 de juny de 2016 2 Agra¨ıments En primer lloc vull agrair als caps del grup, el Dr. Jaume Farr´esi el Dr. Xavier Par´esl'oportunitat de posar un peu a la ci`encia,iniciar-me en la carrera cient´ıfica,els seus bons consells i l’obtenci´o final d'aquesta tesi. Tamb´evoldria agrair especialment a tots els ADHs amb qui he compartit laboratori, sense vosaltres aix`ono hauria estat possible. Gr`aciesper aguantar tants dies a les fosques! Al Sergio-せんせい, per tot el que m'has ensenyat durant aquests anys, la bona guia, direcci´o, les inacabables correccions, per ensenyar-me a ser cr´ıticamb la meva pr`opiafeina i intentar sempre ser rigor´osi auto exigent tant amb els experiments com amb la redacci´oi preparaci´ode les figures (repassar cada figura mil cops buscant defectes abans de donar-la per bona ja s'ha convertit en costum) i tots i cada un dels inacabables \Podries..."que tant hem trobat a faltar en la `epoca final. -

The Role of AKR1B10 in Physiology and Pathophysiology
H OH metabolites OH Review The Role of AKR1B10 in Physiology and Pathophysiology Satoshi Endo 1,* , Toshiyuki Matsunaga 2 and Toru Nishinaka 3 1 Laboratory of Biochemistry, Gifu Pharmaceutical University, Gifu 501-1196, Japan 2 Education Center of Green Pharmaceutical Sciences, Gifu Pharmaceutical University, Gifu 502-8585, Japan; [email protected] 3 Laboratory of Biochemistry, Faculty of Pharmacy, Osaka Ohtani University, Tondabayashi 584-8540, Osaka, Japan; [email protected] * Correspondence: [email protected] Abstract: AKR1B10 is a human nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reductase belonging to the aldo-keto reductase (AKR) 1B subfamily. It catalyzes the reduction of aldehydes, some ketones and quinones, and interacts with acetyl-CoA carboxylase and heat shock protein 90α. The enzyme is highly expressed in epithelial cells of the stomach and intestine, but down-regulated in gastrointestinal cancers and inflammatory bowel diseases. In contrast, AKR1B10 expression is low in other tissues, where the enzyme is upregulated in cancers, as well as in non- alcoholic fatty liver disease and several skin diseases. In addition, the enzyme’s expression is elevated in cancer cells resistant to clinical anti-cancer drugs. Thus, growing evidence supports AKR1B10 as a potential target for diagnosing and treating these diseases. Herein, we reviewed the literature on the roles of AKR1B10 in a healthy gastrointestinal tract, the development and progression of cancers and acquired chemoresistance, in addition to its gene regulation, functions, and inhibitors. Keywords: aldo-keto reductases; AKR1B10; biomarkers Citation: Endo, S.; Matsunaga, T.; Nishinaka, T. The Role of AKR1B10 in 1. Introduction Physiology and Pathophysiology. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur Et Al
US 2012026.6329A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0266329 A1 Mathur et al. (43) Pub. Date: Oct. 18, 2012 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/10 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/24 (2006.01) CI2N 9/02 (2006.01) (75) Inventors: Eric J. Mathur, Carlsbad, CA CI2N 9/06 (2006.01) (US); Cathy Chang, San Marcos, CI2P 2L/02 (2006.01) CA (US) CI2O I/04 (2006.01) CI2N 9/96 (2006.01) (73) Assignee: BP Corporation North America CI2N 5/82 (2006.01) Inc., Houston, TX (US) CI2N 15/53 (2006.01) CI2N IS/54 (2006.01) CI2N 15/57 2006.O1 (22) Filed: Feb. 20, 2012 CI2N IS/60 308: Related U.S. Application Data EN f :08: (62) Division of application No. 1 1/817,403, filed on May AOIH 5/00 (2006.01) 7, 2008, now Pat. No. 8,119,385, filed as application AOIH 5/10 (2006.01) No. PCT/US2006/007642 on Mar. 3, 2006. C07K I4/00 (2006.01) CI2N IS/II (2006.01) (60) Provisional application No. 60/658,984, filed on Mar. AOIH I/06 (2006.01) 4, 2005. CI2N 15/63 (2006.01) Publication Classification (52) U.S. Cl. ................... 800/293; 435/320.1; 435/252.3: 435/325; 435/254.11: 435/254.2:435/348; (51) Int. Cl. 435/419; 435/195; 435/196; 435/198: 435/233; CI2N 15/52 (2006.01) 435/201:435/232; 435/208; 435/227; 435/193; CI2N 15/85 (2006.01) 435/200; 435/189: 435/191: 435/69.1; 435/34; CI2N 5/86 (2006.01) 435/188:536/23.2; 435/468; 800/298; 800/320; CI2N 15/867 (2006.01) 800/317.2: 800/317.4: 800/320.3: 800/306; CI2N 5/864 (2006.01) 800/312 800/320.2: 800/317.3; 800/322; CI2N 5/8 (2006.01) 800/320.1; 530/350, 536/23.1: 800/278; 800/294 CI2N I/2 (2006.01) CI2N 5/10 (2006.01) (57) ABSTRACT CI2N L/15 (2006.01) CI2N I/19 (2006.01) The invention provides polypeptides, including enzymes, CI2N 9/14 (2006.01) structural proteins and binding proteins, polynucleotides CI2N 9/16 (2006.01) encoding these polypeptides, and methods of making and CI2N 9/20 (2006.01) using these polynucleotides and polypeptides. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -
Table Legend
Bin 1 Bins 2 & 3 Bins 4 & 5 Bins 6 & 7 Bin 8 Bin 9 Bin 10 Accession Accession Accession Accession Accession Accession Accession Gene Name Gene Name Gene Name Gene Name Gene Name Gene Name Gene Name Number Number Number Number Number Number Number U70270 Mud-4 U70268 Mud-7 H31839 EST (Bak1) BCL2-antagonist/killer 1 U77829 Gas-5 growth arrest homolog AF025670 Caspase 6 (CASP6); MCH2 AA800243 EST (CIDE-A) Cell death inducing DNA fragmenta AA859938 EST (Bnip23l) BCL2/adenovirus E1B 19 kDa-inte AA818072 EST Bcl-2 associated death agonist AJ006971 DAP-like kinase U77829 Gas-5 growth arrest homolog U77933 Nedd2/Ich-1 M64733 Testosterone-repressed prostate message 2 AA891842 EST Death rec 6, (TNFRSF21) tumor necrosis fa U72995 MAP-kinase activating death domain AA891933 EST Smac protein, mitochondrial precursor, (seconU94340 ADP-ribosyltransferase (NAD+; poly (ADP-ribose) U47315 R2 cerebellum DDRT-T-PCR AA875455 EST Wig1, p53-activated gene 608 U05989 Par-4 induced by effectors of apoptosis AA874794 EST (TNFRSF16) NGF receptor associated protein AA848545 EST Programmed cell death 10, TF-1 cell apoptosi AA874794 EST (TNFRSF16) NGF receptor associated protein U49930 Caspase 3, apoptosis related cysteine protease (IC U49930 Caspase 3, apoptosis related cysteine protease (IC U84410 Caspase 3, apoptosis related cysteine protease (IC AA891591 EST Programmed cell death 8 (apoptosis-inducing AA892388 EST Death-associated kinase 2 AF030358 Chemokine CX3C AA818894 EST (Pgsg) proteoglycan peptide core protein S79676 IL-1 Beta converting enzyme U14647 Interleukin