Mechanisms of Ketogenic Diet Action
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Index Vol. 12-15
353 INDEX VOL. 12-15 Die Stichworte des Sachregisters sind in der jeweiligen Sprache der einzelnen Beitrage aufgefiihrt. Les termes repris dans la Table des matieres sont donnes selon la langue dans laquelle l'ouvrage est ecrit. The references of the Subject Index are given in the language of the respective contribution. 14 AAG (Alpha-acid glycoprotein) 120 14 Adenosine 108 12 Abortion 151 12 Adenosine-phosphate 311 13 Abscisin 12, 46, 66 13 Adenosine-5'-phosphosulfate 148 14 Absorbierbarkeit 317 13 Adenosine triphosphate 358 14 Absorption 309, 350 15 S-Adenosylmethionine 261 13 Absorption of drugs 139 13 Adipaenin (Spasmolytin) 318 14 - 15 12 Adrenal atrophy 96 14 Absorptionsgeschwindigkeit 300, 306 14 - 163, 164 14 Absorptionsquote 324 13 Adrenal gland 362 14 ACAI (Anticorticocatabolic activity in 12 Adrenalin(e) 319 dex) 145 14 - 209, 210 12 Acalo 197 15 - 161 13 Aceclidine (3-Acetoxyquinuclidine) 307, 13 {i-Adrenergic blockers 119 308, 310, 311, 330, 332 13 Adrenergic-blocking activity 56 13 Acedapsone 193,195,197 14 O(-Adrenergic blocking drugs 36, 37, 43 13 Aceperone (Acetabutone) 121 14 {i-Adrenergic blocking drugs 38 12 Acepromazin (Plegizil) 200 14 Adrenergic drugs 90 15 Acetanilid 156 12 Adrenocorticosteroids 14, 30 15 Acetazolamide 219 12 Adrenocorticotropic hormone (ACTH) 13 Acetoacetyl-coenzyme A 258 16,30,155 12 Acetohexamide 16 14 - 149,153,163,165,167,171 15 1-Acetoxy-8-aminooctahydroindolizin 15 Adrenocorticotropin (ACTH) 216 (Slaframin) 168 14 Adrenosterone 153 13 4-Acetoxy-1-azabicyclo(3, 2, 2)-nonane 12 Adreson 252 -

(19) United States (12) Patent Application Publication (10) Pub
US 20130289061A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0289061 A1 Bhide et al. (43) Pub. Date: Oct. 31, 2013 (54) METHODS AND COMPOSITIONS TO Publication Classi?cation PREVENT ADDICTION (51) Int. Cl. (71) Applicant: The General Hospital Corporation, A61K 31/485 (2006-01) Boston’ MA (Us) A61K 31/4458 (2006.01) (52) U.S. Cl. (72) Inventors: Pradeep G. Bhide; Peabody, MA (US); CPC """"" " A61K31/485 (201301); ‘4161223011? Jmm‘“ Zhu’ Ansm’ MA. (Us); USPC ......... .. 514/282; 514/317; 514/654; 514/618; Thomas J. Spencer; Carhsle; MA (US); 514/279 Joseph Biederman; Brookline; MA (Us) (57) ABSTRACT Disclosed herein is a method of reducing or preventing the development of aversion to a CNS stimulant in a subject (21) App1_ NO_; 13/924,815 comprising; administering a therapeutic amount of the neu rological stimulant and administering an antagonist of the kappa opioid receptor; to thereby reduce or prevent the devel - . opment of aversion to the CNS stimulant in the subject. Also (22) Flled' Jun‘ 24’ 2013 disclosed is a method of reducing or preventing the develop ment of addiction to a CNS stimulant in a subj ect; comprising; _ _ administering the CNS stimulant and administering a mu Related U‘s‘ Apphcatlon Data opioid receptor antagonist to thereby reduce or prevent the (63) Continuation of application NO 13/389,959, ?led on development of addiction to the CNS stimulant in the subject. Apt 27’ 2012’ ?led as application NO_ PCT/US2010/ Also disclosed are pharmaceutical compositions comprising 045486 on Aug' 13 2010' a central nervous system stimulant and an opioid receptor ’ antagonist. -

The Effect of Diazepam on Neonatal Seizure: in Vivo 31Pand 'H NMR Study1
003 1-3998/89/2501-0027$02.00/0 PEDIATRIC RESEARCH Vol. 25, No. 1, 1989 Copyright O 1989 International Pediatric Research Foundation, Inc. Printed in U.S.A. The Effect of Diazepam on Neonatal Seizure: In Vivo 31Pand 'H NMR Study1 RICHARD S. K. YOUNG, BENJAMIN CHEN, OGNEN A. C. PETROFF, JOHN C. GORE, BARRETT E. COWAN, EDWARD J. NOVOTNY, JR., MABEL WONG, AND KAYE ZUCKERMAN Departments of Pediatrics [R.S.K. Y., M. W., K.Z.], Neurology [R.S.K. Y., O.A.C.P., E.J.N.], and Diagnostic Radiology [B.C., J.C.G.], Yale University School of Medicine, New Haven, Connecticut 06510-8064, and Department of Medicine [B.E.C.] Stanford University Medical School, Stanford, California 94305 ABSTRACI'. It is assumed that when anticonvulsants this study, four types of investigations were performed: in vivo arrest seizure, there is rapid return of brain high energy 31PNMR to follow brain high energy phosphate metabolism and phosphates and brain lactate to control values. To test this brain pH; in vivo 'H NMR to measure brain lactate; in vitro 'H hypothesis, diazepam was administered to neonatal dogs NMR spectroscopy to determine concentrations of brain excita- during flurothyl-induced seizure. In vivo 3'P nuclear mag- tory and inhibitory amino acids; and iodoantipyrine autoradi- netic resonance spectroscopy disclosed that diazepam ography to assess regional cerebral blood flow. quickly arrested electrographic seizure and restored brain phosphocreatine and inorganic phosphate to baseline val- ues. In contrast, in vivo 'H nuclear magnetic resonance MATERIALS AND METHODS spectroscopic measurements showed that arrest of seizure Animal preparation. -

Norepinephrine-Deficient Mice Have Increased Susceptibility to Seizure
The Journal of Neuroscience, December 15, 1999, 19(24):10985–10992 Norepinephrine-Deficient Mice Have Increased Susceptibility to Seizure-Inducing Stimuli Patricia Szot,1,6 David Weinshenker,2,5 Sylvia S. White,1 Carol A. Robbins,3 Nicole C. Rust,2,5 Philip A. Schwartzkroin,3,4 and Richard D. Palmiter2,5 Departments of 1Psychiatry and Behavioral Science, 2Biochemistry, 3Neurological Surgery, and 4Physiology/Biophysics and 5Howard Hughes Medical Institute, University of Washington, Seattle, Washington 98195, and 6GRECC, Puget Sound Health Care System, Seattle, Washington 98108 Several lines of evidence suggest that norepinephrine (NE) can acid. c-Fos mRNA expression in the cortex, hippocampus (CA1 modulate seizure activity. However, the experimental methods and CA3), and amygdala was increased in Dbh 2/2 mice in used in the past cannot exclude the possible role of other association with flurothyl-induced seizures. Enhanced seizure neurotransmitters coreleased with NE from noradrenergic ter- susceptibility to flurothyl and increased seizure-induced c-fos minals. We have assessed the seizure susceptibility of geneti- mRNA expression were reversed by pretreatment with L-threo- cally engineered mice that lack NE. Seizure susceptibility was 3,4-dihydroxyphenylserine, which partially restores the NE con- determined in the dopamine b-hydroxylase null mutant (Dbh tent in Dbh 2/2 mice. These genetically engineered mice 2/2) mouse using four different convulsant stimuli: 2,2,2- confirm unambiguously the potent effects of the noradrenergic trifluroethyl ether (flurothyl), pentylenetetrazol (PTZ), kainic acid, system in modulating epileptogenicity and illustrate the unique and high-decibel sound. Dbh 2/2 mice demonstrated en- opportunity offered by Dbh 2/2 mice for elucidating the path- hanced susceptibility (i.e., lower threshold) compared with lit- ways through which NE can regulate seizure activity. -

Control of Epileptic Seizures by the Basal Ganglia: Clinical and Experimental Approaches Feddersen Berend
Control of epileptic seizures by the basal ganglia: clinical and experimental approaches Feddersen Berend To cite this version: Feddersen Berend. Control of epileptic seizures by the basal ganglia: clinical and experimental ap- proaches. Neurons and Cognition [q-bio.NC]. Université Joseph-Fourier - Grenoble I, 2009. English. tel-00413972 HAL Id: tel-00413972 https://tel.archives-ouvertes.fr/tel-00413972 Submitted on 7 Sep 2009 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Ecole Doctorale Chimie et Sciences du Vivant UNIVERSITE JOSEPH FOURIER GRENOBLE Thèse Neuroscience - Neurobiologie Berend Feddersen Control of epileptic seizures by the basal ganglia: clinical and experimental approaches Soutenue publiquement le: 10.07.2009 Membres du jury : Franck Semah (Rapporteur 1) Stephane Charpier (Rapporteur 2) Philippe Kahane Soheyl Noachtar Antoine Depaulis (Directeur de thèse) Colin Deransart 1 ACKNOWLEDGEMENTS Many fantastic people were inolved in this thesis, whom I want to thank deeply for all their help and support. I want to thank my supervisors Soheyl Noachtar, Antoine Depaulis and Colin Deransart for all their help and fruitfull discussions in every kind of situation. Sohyel Nochtar teached me in a perfect structured manner clinical epileptology and gave me always all the support I needed, especially for my stay in Grenoble. -

Progressive NKCC1-Dependent Neuronal Chloride Accumulation During Neonatal Seizures
The Journal of Neuroscience, September 1, 2010 • 30(35):11745–11761 • 11745 Neurobiology of Disease Progressive NKCC1-Dependent Neuronal Chloride Accumulation during Neonatal Seizures Volodymyr I. Dzhala,1* Kishore V. Kuchibhotla,1,3* Joseph C. Glykys,1 Kristopher T. Kahle,2 Waldemar B. Swiercz,1 Guoping Feng,4 Thomas Kuner,4 George J. Augustine,4 Brian J. Bacskai,1 and Kevin J. Staley1 Departments of 1Neurology and 2Neurosurgery, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts 02114, 3Program in Biophysics, Harvard University, Cambridge, Massachusetts 02138, and 4Department of Neurobiology, Duke University Medical Center, Durham, North Carolina 27710 Seizures induce excitatory shifts in the reversal potential for GABAA-receptor-mediated responses, which may contribute to the intrac- tability of electro-encephalographic seizures and preclude the efficacy of widely used GABAergic anticonvulsants such as phenobarbital. We now report that, in intact hippocampi prepared from neonatal rats and transgenic mice expressing Clomeleon, recurrent seizures Ϫ progressively increase the intracellular chloride concentration ([Cl ]i ) assayed by Clomeleon imaging and invert the net effect of GABAA receptor activation from inhibition to excitation assayed by the frequency of action potentials and intracellular Ca 2ϩ transients. These changes correlate with increasing frequency of seizure-like events and reduction in phenobarbital efficacy. The Na ϩ–K ϩ–2Cl Ϫ (NKCC1) cotransporter blocker bumetanide inhibited seizure-induced neuronal Cl Ϫ accumulation and the consequent facilitation of recurrent Ϫ seizures. Our results demonstrate a novel mechanism by which seizure activity leads to [Cl ]i accumulation, thereby increasing the probability of subsequent seizures. This provides a potential mechanism for the early crescendo phase of neonatal seizures. -
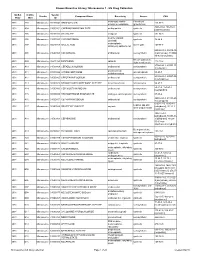
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Bypassing the Blood–Brain Barrier: Direct Intracranial Drug Delivery in Epilepsies
pharmaceutics Review Bypassing the Blood–Brain Barrier: Direct Intracranial Drug Delivery in Epilepsies Manuela Gernert 1,2,* and Malte Feja 1,2 1 Department of Pharmacology, Toxicology, and Pharmacy, University of Veterinary Medicine Hannover, Bünteweg 17, D-30559 Hannover, Germany; [email protected] 2 Center for Systems Neuroscience, D-30559 Hannover, Germany * Correspondence: [email protected]; Tel.: +49-(0)511-953-8527 Received: 30 October 2020; Accepted: 21 November 2020; Published: 24 November 2020 Abstract: Epilepsies are common chronic neurological diseases characterized by recurrent unprovoked seizures of central origin. The mainstay of treatment involves symptomatic suppression of seizures with systemically applied antiseizure drugs (ASDs). Systemic pharmacotherapies for epilepsies are facing two main challenges. First, adverse effects from (often life-long) systemic drug treatment are common, and second, about one-third of patients with epilepsy have seizures refractory to systemic pharmacotherapy. Especially the drug resistance in epilepsies remains an unmet clinical need despite the recent introduction of new ASDs. Apart from other hypotheses, epilepsy-induced alterations of the blood–brain barrier (BBB) are thought to prevent ASDs from entering the brain parenchyma in necessary amounts, thereby being involved in causing drug-resistant epilepsy. Although an invasive procedure, bypassing the BBB by targeted intracranial drug delivery is an attractive approach to circumvent BBB-associated drug resistance mechanisms and to lower the risk of systemic and neurologic adverse effects. Additionally, it offers the possibility of reaching higher local drug concentrations in appropriate target regions while minimizing them in other brain or peripheral areas, as well as using otherwise toxic drugs not suitable for systemic administration. -

Role of Hormones and Neurosteroids in Epileptogenesis
REVIEW ARTICLE published: 31 July 2013 doi: 10.3389/fncel.2013.00115 Role of hormones and neurosteroids in epileptogenesis Doodipala Samba Reddy* Department of Neuroscience and Experimental Therapeutics, College of Medicine, Texas A&M University Health Science Center, Bryan, TX, USA Edited by: This article describes the emerging evidence of hormonal influence on epileptogenesis, Roberto Di Maio, University of which is a process whereby a brain becomes progressively epileptic due to an initial Pittsburgh, USA precipitating event of diverse origin such as brain injury, stroke, infection, or prolonged Reviewed by: seizures. The molecular mechanisms underlying the development of epilepsy are poorly Corette J. Wierenga, Utrecht University, Netherlands understood. Neuroinflammation and neurodegeneration appear to trigger epileptogenesis. Jafri Malin Abdullah, Universiti Sains There is an intense search for drugs that truly prevent the development of epilepsy in Malaysia, Malaysia people at risk. Hormones play an important role in children and adults with epilepsy. *Correspondence: Corticosteroids, progesterone, estrogens, and neurosteroids have been shown to affect D. Samba Reddy, Department of seizure activity in animal models and in clinical studies. However, the impact of hormones Neuroscience and Experimental Therapeutics, College of Medicine, on epileptogenesis has not been investigated widely. There is emerging new evidence Texas A&M University Health Science that progesterone, neurosteroids, and endogenous hormones may play a role in regulating Center, 8447 State Highway 47, the epileptogenesis. Corticosterone has excitatory effects and triggers epileptogenesis MREB Building, Bryan, TX 77807, USA in animal models. Progesterone has disease-modifying activity in epileptogenic models. e-mail: [email protected] The antiepileptogenic effect of progesterone has been attributed to its conversion to neurosteroids, which binds to GABA-A receptors and enhances phasic and tonic inhibition in the brain. -

Experimental Epilepsy: Developmental Aspects
BASIC MECHANISMS 01 CHILDHOOD EPILEPSY N Experimental epilepsy: developmental aspects SOLOMON L. MOSHE, MD; ELLEN F. SPERBER, PHD; LUCY L. BROWN, PHD; ANN TEMPEL, PHD; AND JOHN N.D. WURPEL PHD PIDEMIOLOGIC studies indicate that in hu- that electrographic seizure activity propagates in the mans, seizures occur more frequently early in SN.22-23 Autoradiographic studies of 2-deoxyglucose life.1'2 During the first few years some of the (DG) indicate that during a seizure there is an increase seizure types are age-specific3-4 and difficult to in DG uptake in the SN.15'24-27 Furthermore, informa- Econtrol with conventional therapies.5 The high inci- tion has accumulated suggesting that in adult rats, the dence of seizures cannot be attributed only to the gamma-aminobutyric-acid (GABA)-sensitive substan- greater number of insults that may occur during the tia nigra pars reticulata (SNR) neurons may be respon- newborn period and early infancy. Experimental evi- sible, in part, for the termination of seizures. Infusions dence suggests that the immature central nervous of GABA agonists (such as muscimol) into the SNR system (CNS) is more susceptible to seizures than its can suppress seizures.20'21,28-30 SN lesions20-31 or elec- mature counterpart,6-14 at least during a specific stage trical stimulation32'33 can also suppress seizures; the of development. Ontogenetic seizure studies have dem- effect of electrical stimulation can be reversed by onstrated that 15-to-18-day-old rat pups are more picrotoxin, a GABA antagonist.32 prone to the development of bilateral, asynchronous These observations suggest that treatments which convulsions and status epilepticus than adult rats decrease the GABAergic output of the SNR can regardless of the seizure model used to induce the attenuate or terminate seizures, probably by disinhibit- 6-10 seizures. -

Non-Pharmacological Interventions for Intractable Epilepsy
Saudi Pharmaceutical Journal 28 (2020) 951–962 Contents lists available at ScienceDirect Saudi Pharmaceutical Journal journal homepage: www.sciencedirect.com Review Non-pharmacological Interventions for Intractable Epilepsy ⇑ Faleh Alqahtani a, , Imran Imran b, Hafsa Pervaiz b, Waseem Ashraf b, Nadia Perveen b, Muhammad Fawad Rasool c, Abdullah F. Alasmari a, Metab Alharbi a, Noreen Samad d, Saleh Abdullah Alqarni a, Salim S. Al-Rejaie a, Mohammed Mufadhe Alanazi a a Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia b Department of Pharmacology, Faculty of Pharmacy, Bahauddin Zakariya University, Multan 60800, Pakistan c Department of Pharmacy Practice, Faculty of Pharmacy, Bahauddin Zakariya University, Multan 60800, Pakistan d Department of Biochemistry, Faculty of Science, Bahauddin Zakariya University, Multan 60800, Pakistan article info abstract Article history: In 30% of epileptic individuals, intractable epilepsy represents a problem for the management of seizures Received 14 May 2020 and severely affects the patient’s quality of life due to pharmacoresistance with commonly used anti- Accepted 23 June 2020 seizure drugs (ASDs). Surgery is not the best option for all resistant patients due to its post-surgical con- Available online 29 June 2020 sequences. Therefore, several alternative or complementary therapies have scientifically proven significant therapeutic potential for the management of seizures in intractable epilepsy patients with Keywords: seizure-free occurrences. -

Neurotechnologies As Weapons in National Intelligence and Defense – an Overview James Giordano, Phd*1-3 and Rachel Wurzman, Phd(C)4
Neurotechnologies as weapons in national intelligence and defense – An overview James Giordano, PhD*1-3 and Rachel Wurzman, PhD(c)4 1. Center for Neurotechnology Studies, Potomac Institute for Policy Studies, Arlington, VA, 22203, USA, 2. Department of Electrical and Computational Engineering, University of New Mexico, Albuquerque, NM, 87131, USA, 3. Oxford Centre for Neuroethics, University of Oxford, UK, Email: [email protected], 4. Interdisciplinary Program in Neuroscience, Georgetown University Medical Center, Washington, DC, 20057, USA. Abstract Advances in neuroscience and neurotechnology have necessitated discussions on the ways that such developments could be used as weapons in contexts of national security, intelligence, and defense. This paper defi nes the concept of neuroweapons, and elucidates operational issues associated with their use to aid informational and strategic intelligence, such as brain-machine interfaces to improve effi ciency in data analysis. As well, exploration of neuropharmacologic, neuromicrobiological, and neurotoxic agents are discussed relevant to their utility in combat scenarios. The limitations of emerging neurotechnologies as weapons are addressed, as both regards practical and operational frameworks, and implications relevant to formulation of ethico-legal guidelines and governance of research, development and potential use. Key words: neuroscience, neurotechnology, neuroweapons, neurosecurity, national defense, biotechnology, weaponry Introduction summarized the state of neuroscience