Effect of Memantine, an Anti-Alzheimer's Drug, on Rodent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

First Xenon-Xenon Collisions in the Lhc
9th International Particle Accelerator Conference IPAC2018, Vancouver, BC, Canada JACoW Publishing ISBN: 978-3-95450-184-7 doi:10.18429/JACoW-IPAC2018-MOPMF039 FIRST XENON-XENON COLLISIONS IN THE LHC M. Schaumann∗, R. Alemany-Fernandez, P. Baudrenghien, T. Bohl, C. Bracco, R. Bruce, N. Fuster-Martinez, M. A. Jebramcik, J.M. Jowett, T. Mertens, D. Mirarchi, S. Redaelli, B. Salvachua, M. Solfaroli, H. Timko, J. Wenninger, CERN, Geneva, Switzerland Abstract EXECUTION OF THE RUN In 2017, the CERN accelerator complex once again demonstrated its flexibility by producing beams of a new A total of about 18 h of LHC time were taken for the Xe ion species, xenon, that were successfully injected into LHC. collision run. The schedule was designed to include set-up, On 12 October, collisions of fully stripped xenon nuclei first injection, validation and physics data taking. A further 12 h were devoted to experiments on crystal collimation, de- were recorded for the first time in the LHC√ at a centre-of- scribed elsewhere [3]. This tight schedule was only feasible mass energy per colliding nucleon pair of sNN = 5.44 TeV. Physics data taking started 9.5hafter the first injection of by drastically reducing the complexity of the set-up follow- xenon beams and lasted a total of 6h. The integrated lumi- ing the model of the p–Pb pilot run in September 2012 [4,5] nosity delivered to the four LHC experiments was sufficient when first injection, validation and physics data-taking were that new physics results can be expected soon. We provide achieved within a single fill. -

XENON X-1100 High-Intensity Pulsed Light System
XENON X-1100 High-Intensity Pulsed Light System The low cost R&D tool for investigating the applicability of Pulsed Light for new and emerging applications. The tool that researchers, scientists and engineers have been looking for is here. An easy-to-use photonic system developed by XENON that will open new doors and lead to new discoveries. The power of Pulsed Light In virtually all disciplines of science and technology there are applications where precise delivery of energy is demanded. One less studied energy delivery mechanism is the use of high-intensity pulsed light. At present the most prevalent example of this is in the use of lasers. However, the point source, coherent and single wavelength nature of lasers, are often not suited to the majority of challenges facing many industries. In these situations, it is crucial to have a broad spectra source so as not to be restricted in choosing materials with specific absorption characteristics. Because XENON sources generate light which extends from the deep UV to IR and often with peak pulse powers measured in megawatts, the ability of these sources to perform tasks such as breaking chemical bonds, sintering, ablating or sterilizing is highly realizable. The high peak powers generated by XENON’s production level systems are possible by tightly controlling the pulse durations measured from a few microseconds to milliseconds. Until now there was no practical method of generating this intense pulsed light in a low-cost R&D tool that was safe, compact and versatile. XENON has developed a system to address this challenge based on over 50 years of exper- tise in the Pulsed Light industry. -

Pharmacology – Inhalant Anesthetics
Pharmacology- Inhalant Anesthetics Lyon Lee DVM PhD DACVA Introduction • Maintenance of general anesthesia is primarily carried out using inhalation anesthetics, although intravenous anesthetics may be used for short procedures. • Inhalation anesthetics provide quicker changes of anesthetic depth than injectable anesthetics, and reversal of central nervous depression is more readily achieved, explaining for its popularity in prolonged anesthesia (less risk of overdosing, less accumulation and quicker recovery) (see table 1) Table 1. Comparison of inhalant and injectable anesthetics Inhalant Technique Injectable Technique Expensive Equipment Cheap (needles, syringes) Patent Airway and high O2 Not necessarily Better control of anesthetic depth Once given, suffer the consequences Ease of elimination (ventilation) Only through metabolism & Excretion Pollution No • Commonly administered inhalant anesthetics include volatile liquids such as isoflurane, halothane, sevoflurane and desflurane, and inorganic gas, nitrous oxide (N2O). Except N2O, these volatile anesthetics are chemically ‘halogenated hydrocarbons’ and all are closely related. • Physical characteristics of volatile anesthetics govern their clinical effects and practicality associated with their use. Table 2. Physical characteristics of some volatile anesthetic agents. (MAC is for man) Name partition coefficient. boiling point MAC % blood /gas oil/gas (deg=C) Nitrous oxide 0.47 1.4 -89 105 Cyclopropane 0.55 11.5 -34 9.2 Halothane 2.4 220 50.2 0.75 Methoxyflurane 11.0 950 104.7 0.2 Enflurane 1.9 98 56.5 1.68 Isoflurane 1.4 97 48.5 1.15 Sevoflurane 0.6 53 58.5 2.5 Desflurane 0.42 18.7 25 5.72 Diethyl ether 12 65 34.6 1.92 Chloroform 8 400 61.2 0.77 Trichloroethylene 9 714 86.7 0.23 • The volatile anesthetics are administered as vapors after their evaporization in devices known as vaporizers. -

Xenon Gas Xe Safety Data Sheet SDS P4677
Xenon Safety Data Sheet P-4677 This SDS conforms to U.S. Code of Federal Regulations 29 CFR 1910.1200, Hazard Communication. Date of issue: 01/01/1979 Revision date: 10/24/2016 Supersedes: 10/02/2014 SECTION: 1. Product and company identification 1.1. Product identifier Product form : Substance Name : Xenon CAS No : 7440-63-3 Formula : Xe Other means of identification : Xenon 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Industrial use. Use as directed. 1.3. Details of the supplier of the safety data sheet Praxair, Inc. 10 Riverview Drive Danbury, CT 06810-6268 - USA T 1-800-772-9247 (1-800-PRAXAIR) - F 1-716-879-2146 www.praxair.com 1.4. Emergency telephone number Emergency number : Onsite Emergency: 1-800-645-4633 CHEMTREC, 24hr/day 7days/week — Within USA: 1-800-424-9300, Outside USA: 001-703-527-3887 (collect calls accepted, Contract 17729) SECTION 2: Hazard identification 2.1. Classification of the substance or mixture GHS-US classification Compressed gas H280 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS04 Signal word (GHS-US) : WARNING Hazard statements (GHS-US) : H280 - CONTAINS GAS UNDER PRESSURE; MAY EXPLODE IF HEATED OSHA-H01 - MAY DISPLACE OXYGEN AND CAUSE RAPID SUFFOCATION Precautionary statements (GHS-US) : P202 - Do not handle until all safety precautions have been read and understood P271+P403 - Use and store only outdoors or in a well-ventilated place CGA-PG05 - Use a back flow preventive device in the piping CGA-PG10 - Use only with equipment rated for cylinder pressure CGA-PG06 - Close valve after each use and when empty CGA-PG02 - Protect from sunlight when ambient temperature exceeds 52°C (125°F) 2.3. -

Lung Ventilation - Xenon
Lung Ventilation - Xenon Special Instructions Patients with severe dyspnea may not be able to tolerate this examination. To be performed only at UNMH. Radiopharmaceutical: Xe-133 gas Dose (Adult/Pediatric): Refer to Nuclear Medicine Dose Chart Route of Administration: Inhalation from a Xe-133 gas delivery system. Patient Preparation: None. Equipment Setup: Gamma Camera: ZOOM as appropriate for small children Collimator: Orbiter: LEAP SPECT-CT/ECAM/Evo/Symbia E: High resolution Computer setup: • Static acquisition • 128 x 128 matrix • Zoom 1.0 (greater for small children)—ensure that the lungs fill a significant portion of the detector for small patients • 10 sec/image for inspiration • 30 sec/image for all other images Patient Positioning: Orbiter: • Seated upright on a stool (preferred) with the detector posterior to the patient. Supine if necessary All others: • Supine Procedure: • If the patient is able to sit upright on a stool for the examination, perform the ventilation portion of the examination on the Orbiter if available. • Before beginning the study, explain the breathing maneuvers required; the patient should rehearse these maneuvers. • Place the mask on the patient and ensure that it fits snugly. • Inspiration (Single-breath) Image: • Instruct the patient to exhale completely and then to take a deep breath as the Xe-133 gas flows in. • The patient should hold his/her breath for 10 sec (if possible) while posterior (and anterior if on a dual-headed camera) imaging is performed. Revised 2017-01 Lung Ventilation - Xenon (continued) • Equilibrium (Rebreathing/Wash-in) Images: • The patient should then breathe normally for 3 images (90 seconds). -
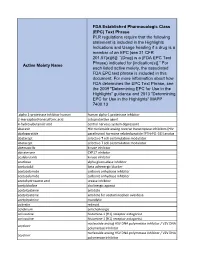
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -

Pharmacokinetics and Pharmacology of Drugs Used in Children
Drug and Fluid Th erapy SECTION II Pharmacokinetics and Pharmacology of Drugs Used CHAPTER 6 in Children Charles J. Coté, Jerrold Lerman, Robert M. Ward, Ralph A. Lugo, and Nishan Goudsouzian Drug Distribution Propofol Protein Binding Ketamine Body Composition Etomidate Metabolism and Excretion Muscle Relaxants Hepatic Blood Flow Succinylcholine Renal Excretion Intermediate-Acting Nondepolarizing Relaxants Pharmacokinetic Principles and Calculations Atracurium First-Order Kinetics Cisatracurium Half-Life Vecuronium First-Order Single-Compartment Kinetics Rocuronium First-Order Multiple-Compartment Kinetics Clinical Implications When Using Short- and Zero-Order Kinetics Intermediate-Acting Relaxants Apparent Volume of Distribution Long-Acting Nondepolarizing Relaxants Repetitive Dosing and Drug Accumulation Pancuronium Steady State Antagonism of Muscle Relaxants Loading Dose General Principles Central Nervous System Effects Suggamadex The Drug Approval Process, the Package Insert, and Relaxants in Special Situations Drug Labeling Opioids Inhalation Anesthetic Agents Morphine Physicochemical Properties Meperidine Pharmacokinetics of Inhaled Anesthetics Hydromorphone Pharmacodynamics of Inhaled Anesthetics Oxycodone Clinical Effects Methadone Nitrous Oxide Fentanyl Environmental Impact Alfentanil Oxygen Sufentanil Intravenous Anesthetic Agents Remifentanil Barbiturates Butorphanol and Nalbuphine 89 A Practice of Anesthesia for Infants and Children Codeine Antiemetics Tramadol Metoclopramide Nonsteroidal Anti-infl ammatory Agents 5-Hydroxytryptamine -

Xenon-Tried-Tested-And-True.Pdf
Tried, tested and true: Why Xenon illumination is the preferred choice over laser phosphor for mainstream cinema Xenon – cinema’s workhorse With 45 years of use and 1.5 million installations worldwide, Christie® Xenolite® lamps are a constant in the cinema industry. Producing impressive light output, accurate color reproduction and boasting 99.999% reliability, this “lightning in a bottle” technology is the dependable workhorse trusted by exhibitors everywhere. Laser phosphor cinema projectors – not to be confused with RGB laser projectors – have been touted as a viable option to replace Xenon-based systems. This has resulted in cinema owners being faced with a decision. Should they continue using Xenon lamps, or make the switch to laser phosphor? Our answer to this is simple. For mainstream theatres, laser phosphor cinema projectors do not provide the performance advantages to justify them as an acceptable option over Xenon-based systems. Brightness over 30,000 hours 20K lumen-class, Xenon versus laser phosphor* 110% (Number of lamps used) (14 fL) 100% 1 2 3 4 5 6 7 8 9 10 11 Xenon 90% 11 fL typical (11 fL) 80% â Below DCI standards Just over 1 year á 70% of operation (based on 10 hours per day) 60% DCI Laser phosphor 7.0 fL typical ed brightness 50% ent of rc quir Pe re 0% 052,800 ,600 8,400 11,200 14,000 16,800 19,600 22,400 25,200 28,000 30,800 Hours of operation * ~20m wide screen * ~1.8 gain screen Laser phosphor – not suitable for cinema laser phosphor projectors. -

Coordination Chemistry of Xenon
Coordination Chemistry of Xenon Paul Griffin Literature Seminar 11/14/19 Situated on the far side of the periodic table, the chemical inertness of the noble gases has led to their widespread applications ranging from gas-discharge lamps and ion engines for space travel to medical applications including as MRI contrast agents and radiotherapy.1 Though all practical applications of the noble gases center around Xe (0), understanding the reactivity and bonding of xenon in higher oxidation states is fundamental to investigating chemistry at the far end of the periodic table. The reactivity of the noble gases, in particular xenon, was first uncovered when Bartlett, who had previously prepared dioxygenyl hexafluoroplatinate (V) (O2PtF6), noted similar ionization energies for molecular oxygen and atomic xenon (a difference of 0.1 eV).2 Mixing of xenon with PtF6 vapor led to the formation of an orange-yellow solid led to the synthesis of the first noble gas compound, xenon hexafluoroplatinate (V) (XePtF6). The chemistry of xenon has evolved significantly since its’ discovery to include an assortment of xenon fluorides, oxides, oxylfluorides, organoxenon3, and metalloxenon compounds such as the tetraxenoaurate (II) cation (Figure 1).4 These species have weak covalent bonding between Xe and the oxygen/fluorine moieties.5 The chemistry of xenon is dominated by the 2+, 4+, and 6+ oxidation states, though Xe (VIII) compounds are known in the form of perxenates and xenon tetroxide. These primarily ionic species, particularly the oxides, oxylfluorides, and fluorides, serve as precursors to noncovalent adducts of xenon. Figure 1. Organoxenon and metalloxenon species Recent work has led to the preparation of xenon adducts with noncovalently interacting ligands such as acetonitrile6a, 6b, ketones6c, sulfoxides6c, and crown ethers6d (Figure 2) capable of weakly coordinating to xenon salts to afford stable species at room temperature. -

Dual Antidepressant Duloxetine Blocks Nicotinic Receptor Currents, Calcium Signals and Exocytosis in Chromaffin Cells Stimulated with Acetylcholine S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/07/13/jpet.118.250969.DC1 1521-0103/367/1/28–39$35.00 https://doi.org/10.1124/jpet.118.250969 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 367:28–39, October 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Dual Antidepressant Duloxetine Blocks Nicotinic Receptor Currents, Calcium Signals and Exocytosis in Chromaffin Cells Stimulated with Acetylcholine s Carmen Nanclares, Isabel Gameiro-Ros, Iago Méndez-López, Carmen Martínez-Ramírez, J. Fernando Padín-Nogueira, Inés Colmena, Andrés M. Baraibar, Luis Gandía,1 and Antonio G. García1 Instituto Teófilo Hernando and Departamento de Farmacología, Facultad de Medicina (C.N., I.G.-R., I.M.-L., C.M.-R., J.F.P.-N., I.C., A.M.B., L.G., A.G.G.) and Instituto de Investigación Sanitaria, Hospital Universitario de La Princesa (A.G.G.), Universidad Downloaded from Autónoma de Madrid, Madrid, Spain; and Departamento de Ciencias Médicas, Facultad de Medicina, Universidad Castilla La Mancha (UCLM), Ciudad Real, Spain (J.F.P.-N.) Received May 28, 2018; accepted July 12, 2018 ABSTRACT jpet.aspetjournals.org The inhibition of nicotinic acetylcholine receptors (nAChRs) partially use dependent. The ACh-elicited membrane depolar- 21 has been proposed as a potential strategy to develop new antide- ization, the elevation of cytosolic calcium ([Ca ]c), and cate- pressant drugs. This is based on the observation that antide- cholamine release in BCCs were also blocked by duloxetine. This pressants that selectively block noradrenaline (NA) or serotonin blockade developed slowly, and the recovery of secretion was (5-HT) reuptake also inhibit nAChRs. -

Pamphlet 155-2 Appendix 1 Data Code Values
Florida Department of Children and Families Substance Abuse and Mental Health Financial and Services Accountability Management System (FASAMS) Pamphlet 155-2 Appendix 1 Data Code Values Last Revision Date: 7/22/2021 Version 13.0 Pamphlet 155-2 Appendix 1, Version 13.0 Last Revision Date 7/22/2021 Page 1 of 71 Table of Contents 1 Children Dependency or Delinquency Status ................................................................................................................. 3 2 County Area .................................................................................................................................................................... 4 3 Covered Service or Project .............................................................................................................................................. 5 4 Discharge Destination ................................................................................................................................................... 18 5 Education Grade Level .................................................................................................................................................. 19 6 Employment Status ....................................................................................................................................................... 19 7 Evaluation Level ............................................................................................................................................................ 20 8 Project Codes -

Sevoflurane, Desflurane, and Xenon New Inhaled Anesthetics in Veterinary Medicine
Ciência Rural, Santa Maria, v.31, n.1, p.177-183, 2001 177 ISSN 0103-8478 SEVOFLURANE, DESFLURANE, AND XENON NEW INHALED ANESTHETICS IN VETERINARY MEDICINE SEVOFLURANO, DESFLURANO E XENÔNIO NOVOS ANESTÉSICOS INALATÓRIOS EM MEDICINA VETERINÁRIA Cláudio Correa Natalini 1 - REVIEW - SUMMARY relação ao uso clínico e a segurança do uso desses novos fármacos em animais. A literatura disponível sobre o uso desses Inhalation anesthesia is widely used in veterinary anestésicos em animais está revisada neste artigo. medicine. New inhalation anesthetics that present less untoward effects, are more potent and produce a safe and easily changeable Palavras-chave: anestesia inalatória, sevoflurano, desflurano, anesthetic plane are desirable over the older agents presently xenônio. available. In this review some of the physical and chemical aspects of inhalation anesthesia is revisited. Because the agents used in inhalation anesthesia are gases or vapors, the physics of vaporization, delivery and administration of these agents should INTRODUCTION be understood. The two new inhalation anesthetics sevoflurane and desflurane, and the new anesthetic gas xenon have been used Inhalation anesthetics are used widely for in human beings for some time. In veterinary medicine there is a the anesthetic management of animals. They are lack of investigation and reports that assure the safety and clinical aspects of using them in animals. The information unique among the anesthetic drugs because they are available on the use of these new agents in animals is revised in administered, and in large part removed from the this article. body, via the lungs. Their popularity arises in part because their pharmacokinetic characteristics favor Key words: inhalation anesthesia, sevoflurane, desflurane, and xenon.