NEURO BOWL Primary Care in Paradise
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia
brain sciences Review Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia Timothy Fullam and Jeffrey Statland * Department of Neurology, University of Kansas Medical Center, Kansas, KS 66160, USA; [email protected] * Correspondence: [email protected] Abstract: Following the exclusion of potentially reversible causes, the differential for those patients presenting with a predominant upper motor neuron syndrome includes primary lateral sclerosis (PLS), hereditary spastic paraplegia (HSP), or upper motor neuron dominant ALS (UMNdALS). Differentiation of these disorders in the early phases of disease remains challenging. While no single clinical or diagnostic tests is specific, there are several developing biomarkers and neuroimaging technologies which may help distinguish PLS from HSP and UMNdALS. Recent consensus diagnostic criteria and use of evolving technologies will allow more precise delineation of PLS from other upper motor neuron disorders and aid in the targeting of potentially disease-modifying therapeutics. Keywords: primary lateral sclerosis; amyotrophic lateral sclerosis; hereditary spastic paraplegia Citation: Fullam, T.; Statland, J. Upper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper 1. Introduction Motor Neuron Dominant Jean-Martin Charcot (1825–1893) and Wilhelm Erb (1840–1921) are credited with first Amyotrophic Lateral Sclerosis, and describing a distinct clinical syndrome of upper motor neuron (UMN) tract degeneration in Hereditary Spastic Paraplegia. Brain isolation with symptoms including spasticity, hyperreflexia, and mild weakness [1,2]. Many Sci. 2021, 11, 611. https:// of the earliest described cases included cases of hereditary spastic paraplegia, amyotrophic doi.org/10.3390/brainsci11050611 lateral sclerosis, and underrecognized structural, infectious, or inflammatory etiologies for upper motor neuron dysfunction which have since become routinely diagnosed with the Academic Editors: P. -

Management of Neurogenic Dysphagia
694 Postgrad Med J 2001;77:694–699 Postgrad Med J: first published as 10.1136/pmj.77.913.694 on 1 November 2001. Downloaded from Management of neurogenic dysphagia A M O Bakheit Dysphagia is common in patients with neuro- of the cerebral cortex, basal ganglia, brain logical disorders. It may result from lesions in stem, cerebellum, and lower cranial nerves may the central or peripheral nervous system as well result in dysphagia. Degeneration of the as from diseases of muscle and disorders of the myenteric ganglion cells in the oesophagus, neuromuscular junction. Drugs that are com- muscle diseases and disorders of neuromusc- monly used in the management of neurological ular transmission, for example myasthenia conditions may also precipitate or aggravate gravis and Eaton-Lambers syndrome, are other swallowing diYculties in some patients. Neuro- less common causes. genic dysphagia often results in serious compli- cations, including pulmonary aspiration, dehy- CEREBRAL CORTEX dration, and malnutrition. These The commonest condition associated with complications are usually preventable if the dysphagia resulting from cortical lesions is stroke. Acute stroke is complicated by dys- dysphagia is recognised early and managed 1 appropriately. phagia in about 25%–42% of all cases. Dysphagia in these patients is usually associ- Physiological mechanisms of neurogenic ated with hemiplegia due to lesions of the brain stem or the involvement of one or both dysphagia The act of swallowing may be viewed as three hemispheres. However, on rare occasions, dys- discrete but inter-related physiological stages: phagia may be the sole manifestation of a cer- the oral, pharyngeal, and oesophageal phases. -

Juvenile Huntington's Disease: a Case of Paternal Transmission With
ISSN: 2378-3656 Agostinho et al. Clin Med Rev Case Rep 2019, 6:253 DOI: 10.23937/2378-3656/1410253 Volume 6 | Issue 1 Clinical Medical Reviews Open Access and Case Reports BRIEF REPORT Juvenile Huntington’s Disease: A Case of Paternal Transmission with an Uncommon CAG Expansion Luciana de Andrade Agostinho1,2,3*, Luiz Felipe Vasconcellos4, Victor Calil da Silveira4, Thays Apolinário1, Michele da Silva Gonçalves6,7, Mariana Spitz8 and Carmen Lúcia Antão Paiva1,5 1Programa de Pós-Graduação em de Neurologia, Universidade Federal do Estado do Rio de Janeiro, Rio de Janeiro, Brazil 2Centro Universitário Faminas, UNIFAMINAS, Muriaé, Brazil 3Hospital do Câncer de Muriaé - Fundação Cristiano Varella, Muriaé, Brazil 4Instituto de Neurologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil 5Departamento de Genética e Biologia Molecular, Universidade Federal do Estado do Rio de Janeiro, Brazil Check for updates 6Laboratório Hermes Pardini, Belo Horizonte, Minas Gerais, Brazil 7Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil 8Serviço de Neurologia, Universidade do Estado do Rio de Janeiro, Rio de Janeiro, Brazil *Corresponding author: Luciana de Andrade Agostinho, Programa de Pós-Graduação em de Neurologia, Universidade Federal do Estado do Rio de Janeiro; Centro Universitário Faminas, UNIFAMINAS; Hospital do Câncer de Muriaé - Fundação Cristiano Varella; Rua Manoel Francisco de Assis, 732, Muriaé (MG), CEP 36880000, Brazil, Tel: +55-32-998181209 Abstract Keywords Background: Juvenile HD (JHD) is the result of genetic CAG repeats, Juvenile Huntington’s disease, UHDRS, An- anticipation that occurs due to instability of CAG expanded ticipation alleles (HTT gene) when passed to the next generation, resulting in earlier onset of clinical manifestations in successive generations. -

ICD9 & ICD10 Neuromuscular Codes
ICD-9-CM and ICD-10-CM NEUROMUSCULAR DIAGNOSIS CODES ICD-9-CM ICD-10-CM Focal Neuropathy Mononeuropathy G56.00 Carpal tunnel syndrome, unspecified Carpal tunnel syndrome 354.00 G56.00 upper limb Other lesions of median nerve, Other median nerve lesion 354.10 G56.10 unspecified upper limb Lesion of ulnar nerve, unspecified Lesion of ulnar nerve 354.20 G56.20 upper limb Lesion of radial nerve, unspecified Lesion of radial nerve 354.30 G56.30 upper limb Lesion of sciatic nerve, unspecified Sciatic nerve lesion (Piriformis syndrome) 355.00 G57.00 lower limb Meralgia paresthetica, unspecified Meralgia paresthetica 355.10 G57.10 lower limb Lesion of lateral popiteal nerve, Peroneal nerve (lesion of lateral popiteal nerve) 355.30 G57.30 unspecified lower limb Tarsal tunnel syndrome, unspecified Tarsal tunnel syndrome 355.50 G57.50 lower limb Plexus Brachial plexus lesion 353.00 Brachial plexus disorders G54.0 Brachial neuralgia (or radiculitis NOS) 723.40 Radiculopathy, cervical region M54.12 Radiculopathy, cervicothoracic region M54.13 Thoracic outlet syndrome (Thoracic root Thoracic root disorders, not elsewhere 353.00 G54.3 lesions, not elsewhere classified) classified Lumbosacral plexus lesion 353.10 Lumbosacral plexus disorders G54.1 Neuralgic amyotrophy 353.50 Neuralgic amyotrophy G54.5 Root Cervical radiculopathy (Intervertebral disc Cervical disc disorder with myelopathy, 722.71 M50.00 disorder with myelopathy, cervical region) unspecified cervical region Lumbosacral root lesions (Degeneration of Other intervertebral disc degeneration, -

The Dystonias
LE JOURNAL CANADIEN DES SCIENCES NEUROLOGIQUES SUBJECT REVIEW The Dystonias Edith G. McGeer and Patrick L. McGeer Can. J. Neurol. Sci. 1988; 15: 447-483 Contents This review is intended for both the practitioner and the sci entist. Its purpose is to summarize current knowledge regarding the various forms of dystonia, as well as the pathology known Introduction to produce the syndrome in specialized circumstances. Histopathological and brain imaging studies The low incidence of the disorder, its prolonged course, and the difficulty of accurate diagnosis has precluded the type of Sleep and other physiological studies systematic investigation that is possible with many other disor Chemical pathology ders. Yet such systematic investigation is essential if the myster Brain studies ies surrounding dystonia are to be unravelled and methods of CSF studies treatment improved. Blood studies Dystonia has been defined by the Scientific Advisory Board Urine studies of the Dystonia Medical Research Foundation as a syndrome of Fibroblast studies sustained muscle contraction, frequently causing twisting and Miscellaneous repetitive movements, or abnormal posture. It is a clinical term Therapy and not a disease description. It refers to all anatomical forms, whether they involve generalized musculature or only focal Iatrogenic dystonia groups. Although dystonia appears as part of the syndrome in a Possible animal models of dystonia number of disease states, it is idiopathic dystonia, where inheri Summary tance is a major factor, that has aroused the greatest medical interest. This review emphasizes recent literature and those aspects which may contribute to an understanding of the under 1. INTRODUCTION lying mechanisms of dystonic movement. -

History-Of-Movement-Disorders.Pdf
Comp. by: NJayamalathiProof0000876237 Date:20/11/08 Time:10:08:14 Stage:First Proof File Path://spiina1001z/Womat/Production/PRODENV/0000000001/0000011393/0000000016/ 0000876237.3D Proof by: QC by: ProjectAcronym:BS:FINGER Volume:02133 Handbook of Clinical Neurology, Vol. 95 (3rd series) History of Neurology S. Finger, F. Boller, K.L. Tyler, Editors # 2009 Elsevier B.V. All rights reserved Chapter 33 The history of movement disorders DOUGLAS J. LANSKA* Veterans Affairs Medical Center, Tomah, WI, USA, and University of Wisconsin School of Medicine and Public Health, Madison, WI, USA THE BASAL GANGLIA AND DISORDERS Eduard Hitzig (1838–1907) on the cerebral cortex of dogs OF MOVEMENT (Fritsch and Hitzig, 1870/1960), British physiologist Distinction between cortex, white matter, David Ferrier’s (1843–1928) stimulation and ablation and subcortical nuclei experiments on rabbits, cats, dogs and primates begun in 1873 (Ferrier, 1876), and Jackson’s careful clinical The distinction between cortex, white matter, and sub- and clinical-pathologic studies in people (late 1860s cortical nuclei was appreciated by Andreas Vesalius and early 1870s) that the role of the motor cortex was (1514–1564) and Francisco Piccolomini (1520–1604) in appreciated, so that by 1876 Jackson could consider the the 16th century (Vesalius, 1542; Piccolomini, 1630; “motor centers in Hitzig and Ferrier’s region ...higher Goetz et al., 2001a), and a century later British physician in degree of evolution that the corpus striatum” Thomas Willis (1621–1675) implicated the corpus -

Deep Brain Stimulation in the Treatment of Dyskinesia and Dystonia
Neurosurg Focus 17 (1):E2, 2004, Click here to return to Table of Contents Deep brain stimulation in the treatment of dyskinesia and dystonia HIROKI TODA, M.D., PH.D., CLEMENT HAMANI, M.D., PH.D., AND ANDRES LOZANO, M.D., PH.D., F.R.C.S.(C) Division of Neurosurgery, Department of Surgery, Toronto Western Hospital, University of Toronto, Ontario, Cananda Deep brain stimulation (DBS) has become a mainstay of treatment for patients with movement disorders. This modality is directed at modulating pathological activity within basal ganglia output structures by stimulating some of their nuclei, such as the subthalamic nucleus (STN) and the globus pallidus internus (GPi), without making permanent lesions. With the accumulation of experience, indications for the use of DBS have become clearer and the effective- ness and limitations of this form of therapy in different clinical conditions have been better appreciated. In this review the authors discuss the efficacy of DBS in the treatment of dystonia and levodopa-induced dyskinesias. The use of DBS of the STN and GPi is very effective for the treatment of movement disorders induced by levodopa. The relative ben- efits of using the GPi as opposed to the STN as a target are still being investigated. Bilateral GPi stimulation is gain- ing importance in the therapeutic armamentarium for the treatment of dystonia. The DYT1 forms of generalized dys- tonia and cervical dystonias respond to DBS better than secondary dystonia does. Discrimination between the diverse forms of dystonia and a better understanding of the pathophysiological features of this condition will serve as a plat- form for improved outcomes. -

THE SYNDROMES of the ARTERIES of the BRAIN and SPINAL CORD Part 1 by LESLIE G
65 Postgrad Med J: first published as 10.1136/pgmj.29.328.65 on 1 February 1953. Downloaded from THE SYNDROMES OF THE ARTERIES OF THE BRAIN AND SPINAL CORD Part 1 By LESLIE G. KILOH, M.D., M.R.C.P., D.P.M. First Assistant in the Joint Department of Psychological Medicine, Royal Victoria Infirmary and University of Durham Introduction general circulatory efficiency at the time of the Little interest is shown at present in the syn- catastrophe is of the greatest importance. An dromes of the arteries of the brain and spinal cord, occlusion, producing little effect in the presence of and this perhaps is related to the tendency to a normal blood pressure, may cause widespread minimize the importance of cerebral localization. pathological changes if hypotension co-exists. Yet these syndromes are far from being fully A marked discrepancy has been noted between elucidated and understood. It must be admitted the effects of ligation and of spontaneous throm- that in many cases precise localization is often of bosis of an artery. The former seldom produces little more than academic interest and ill effects whilst the latter frequently determines provides Protected by copyright. nothing but a measure of personal satisfaction to infarction. This is probably due to the tendency the physician. But it is worth recalling that the of a spontaneous thrombus to extend along the detailed study of the distribution of the bronchi affected vessel, sealing its branches and blocking and of the pathological anatomy of congenital its collateral circulation, and to the fact that the abnormalities of the heart, was similarly neglected arterial disease is so often generalized. -

Part Ii – Neurological Disorders
Part ii – Neurological Disorders CHAPTER 14 MOVEMENT DISORDERS AND MOTOR NEURONE DISEASE Dr William P. Howlett 2012 Kilimanjaro Christian Medical Centre, Moshi, Kilimanjaro, Tanzania BRIC 2012 University of Bergen PO Box 7800 NO-5020 Bergen Norway NEUROLOGY IN AFRICA William Howlett Illustrations: Ellinor Moldeklev Hoff, Department of Photos and Drawings, UiB Cover: Tor Vegard Tobiassen Layout: Christian Bakke, Division of Communication, University of Bergen E JØM RKE IL T M 2 Printed by Bodoni, Bergen, Norway 4 9 1 9 6 Trykksak Copyright © 2012 William Howlett NEUROLOGY IN AFRICA is freely available to download at Bergen Open Research Archive (https://bora.uib.no) www.uib.no/cih/en/resources/neurology-in-africa ISBN 978-82-7453-085-0 Notice/Disclaimer This publication is intended to give accurate information with regard to the subject matter covered. However medical knowledge is constantly changing and information may alter. It is the responsibility of the practitioner to determine the best treatment for the patient and readers are therefore obliged to check and verify information contained within the book. This recommendation is most important with regard to drugs used, their dose, route and duration of administration, indications and contraindications and side effects. The author and the publisher waive any and all liability for damages, injury or death to persons or property incurred, directly or indirectly by this publication. CONTENTS MOVEMENT DISORDERS AND MOTOR NEURONE DISEASE 329 PARKINSON’S DISEASE (PD) � � � � � � � � � � � -
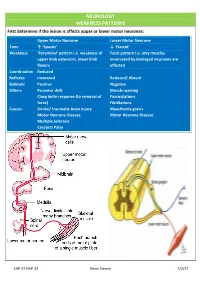
NEUROLOGY WEAKNESS PATTERNS First Determine If the Lesion Is Affects Upper Or Lower Motor Neurones
NEUROLOGY WEAKNESS PATTERNS First determine if the lesion is affects upper or lower motor neurones: Upper Motor Neurone Lower Motor Neurone Tone ↑ ‘Spastic’ ↓ ‘Flaccid’ Weakness ‘Pyramidal’ pattern i.e. weakness of Focal pattern i.e. only muscles upper limb extensors, lower limb innervated by damaged neurones are flexors affected Coordination Reduced Reflexes Increased Reduced/ Absent Babinski Positive Negative Others Pronator drift Muscle wasting Clasp knife response (to removal of Fasciculations force) Fibrillations Causes Stroke/ traumatic brain injury Myasthenia gravis Motor Neurone Disease Motor Neurone Disease Multiple Sclerosis Cerebral Palsy CAP 37 HAP 33 Kevin Gervin 7/2/17 What do we mean by extrapyramidal? Extrapyramidal symptoms Symptoms affecting tracts other than corticospinal and corticobulbar Extrapyramidal tracts don’t travel through the medullary pyramids The system regulates posture and muscle tone so pathology usually leads to movement disorders Most common cause is typical antipsychotics affecting Dopamine (D2) receptors e.g. haloperidol Treatment is anticholinergics e.g. procyclidine Extrapyramidal conditions: Acute dystonic reactions → muscle spasms e.g. neck, jaw, back. Akathisia- feeling of internal restlessness Drug induced Parkinsonism- tremor, rigidity etc. Tardive dyskinesia- involuntary muscle movements of lower face and extremities, often permanent What’s the difference between a Bulbar and Pseudobulbar palsy? Bulbar Pseudobulbar Pathophysiology LMN lesion CN V, VII, IX- XII Disease of corticobulbar -

The Clinical Approach to Movement Disorders Wilson F
REVIEWS The clinical approach to movement disorders Wilson F. Abdo, Bart P. C. van de Warrenburg, David J. Burn, Niall P. Quinn and Bastiaan R. Bloem Abstract | Movement disorders are commonly encountered in the clinic. In this Review, aimed at trainees and general neurologists, we provide a practical step-by-step approach to help clinicians in their ‘pattern recognition’ of movement disorders, as part of a process that ultimately leads to the diagnosis. The key to success is establishing the phenomenology of the clinical syndrome, which is determined from the specific combination of the dominant movement disorder, other abnormal movements in patients presenting with a mixed movement disorder, and a set of associated neurological and non-neurological abnormalities. Definition of the clinical syndrome in this manner should, in turn, result in a differential diagnosis. Sometimes, simple pattern recognition will suffice and lead directly to the diagnosis, but often ancillary investigations, guided by the dominant movement disorder, are required. We illustrate this diagnostic process for the most common types of movement disorder, namely, akinetic –rigid syndromes and the various types of hyperkinetic disorders (myoclonus, chorea, tics, dystonia and tremor). Abdo, W. F. et al. Nat. Rev. Neurol. 6, 29–37 (2010); doi:10.1038/nrneurol.2009.196 1 Continuing Medical Education online 85 years. The prevalence of essential tremor—the most common form of tremor—is 4% in people aged over This activity has been planned and implemented in accordance 40 years, increasing to 14% in people over 65 years of with the Essential Areas and policies of the Accreditation Council age.2,3 The prevalence of tics in school-age children and for Continuing Medical Education through the joint sponsorship of 4 MedscapeCME and Nature Publishing Group. -

Isolated Facial Palsy: a New Lacunar Syndrome
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.47.1.84 on 1 January 1984. Downloaded from Journal ofNeurology, Neurosuirgery, and Psychiatry 1984;47: 84-86 Short report Isolated facial palsy: a new lacunar syndrome CHENYA HUANG, GERALD BROE From the Department ofMedicine, University ofHong Kong, Queen Mary Hospital, Hong Kong and Department ofNeurology, Lidcombe Hospital, Lidcombe, Australia SUMMARY Three cases of sudden isolated upper motor neuron facial palsy and two with associated pseudobulbar palsy have been seen. All were without significant limb weakness. Computed tomography demonstrated small deep infarcts in the internal capsular/corona radiata regions. Pure upper motor neuron facial palsy may be another lacunar syndrome, due to a lesion in the internal capsule or corona radiata. palsies are lower motor neuron lesions. On examination blood pressure was 130/90 mm Hg. He Most facial facial paresis, complete an isolated facial palsy is seen had a left upper motor neuron Occasionally however, and aphagia. Jaw jerk was not increased. Tone, motor neuron in nature. anarthria which appears to be upper power, coordination and sensation were normal; however Protected by copyright. Prior to the availability of CT scan, the site of such the patient veered slightly to the left on walking. CSF lesion was unknown. Some have been ascribed to examination showed 1 mononuclear cell/mm' with normal peripheral facial nerve lesions. ' Puvenendren, Wong biochemistry and serology. EMG of facial muscles showed and Ransome described in 1979 five patients with no abnormality. CT scan showed small low density lesions sudden onset of dysarthria, dysphagia and upper bilaterally in the region of corona radiata adjacent to the motor neuron facial palsy.' EEG and isotope scans basal ganglia (fig 1).