UNIVERSITY of CALIFORNIA, SAN DIEGO the Effects of Cytoskeletal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nervous Tissue
Department of Histology and Embryology Medical faculty KU Bratislava NERVOUS TISSUE RNDr. Mária Csobonyeiová, PhD ([email protected]) Nerve tissue neurons /main cells/ (perikaryon = cell body=soma,dendrites,axon), 4 -150 µm glial cells /supporting cells/ - 10 times more abudant CNS- oligodendrocytes, astrocytes, ependymal cells,microglia PNS - Schwann cells, satelite cells Neuron independentNeuron anatomical and functional unit responsible for: receiving of different types of stimuli transducing them into the nerve impulses conducting them to the nerve centers development – embryonal neuroectoderm Morphology of the neurons Pseudounipolar neuron! (spinal ganglion) Methods used in neurohistology Staining methods: Luxol blue and cresyl violet (nucleus+nucleolus+Nissl body) Luxol blue (myelin sheath) and nuclear red (nucleus + nucleolus+Nissl body) Impregnations according - Holmes – neurons, axon, dendrites - neurofibrils (brown-violet) Golgi – neurons + astrocytes (black) with golden background Cajal – astrocytes (black) with red background Rio del Hortega – microglia (black) with gray-violet background OsO4 - myelin sheath (black), staining for lipids and lipoproteins (myelin) Microglia (phagocytosis) Astrocytes (supporting role, Oligodendrocytes nutrition, healing (formation of myelin of defects - glial sheath) scars, formation of BBB) Ependymal cells (regulation of stable chemical constitution of CSF) CSN Gray matter: White matter: - bodies of neurons, dendrites - myelinated and unmyelinated axons - initial portion -

L03 Neurons to Post.Key
Vert Phys PCB3743 Neurons Fox Chapter 7 pt 1 © T. Houpt, Ph.D. Structure of Vertebrates Two major compartments of the body Peripheral Compartment Everything outside of the brain and spinal cord (heart, lungs, gastrointestinal tract, liver, kidneys, skeletal muscle, skin etc.) Central Nervous System (CNS) • Brain at front of body • Spinal cord running down the back • Protected by skull and vertebra • Sensory receptors clustered in head (vision, hearing, taste, smell) T http://bookdome.com/health/anatomy/Human-Body/Man-Is-A-Vertebrate-Animal.html Vertebrate Central Nervous System: brain & spinal cord cerebellum cerebrum back brainstem Vertebra Skull spinal cord head tail GI tract stomach Vertebrate Central Nervous System: brain & spinal cord cerebellum cerebrum back brainstem spinal cord head tail GI tract stomach Peripheral Nervous System: Neurons and nerve fibers outside the brain and spinal cord back motor neurons sensory ganglion autonomic ganglion head autonomic motor sensory tail nerve nerve nerve GI tract enteric NS stomach Functions of the Nervous System Sensory Motor Integration Detect changes in the environment or in the body via sensory receptors; coordinate responses across the body. Initiate responses via skeletal muscle (somatic nerves for voluntary movement) or via smooth muscle and glands (autonomic nervous system). Neurons (nerve cells) Point to point communication across the body to coordinate responses Integrate electrical and chemical signals at dendrites & cell body; depending on inputs, neuron sends electrical and chemical signal down axon to synapse on target cell. Sensory neurons (afferents) carry sensory information into the CNS Motor neurons (efferents) carry impulses out of CNS to make muscles move or effect target organs (e.g. -

Normal Cells of Cns
NORMAL CELLS OF CNS OBJECTIVES: At the end of this lecture, you should describe the microscopic structure and the function of: 1- Neurons: - Cell body (perikaryon). - Processes: An axon and dendrites. 2- Neuroglia: - Astrocytes. - Oligodendrocytes. - Microglia. - Ependymal cells. Neuron Components: 1. Cell body (Perikaryon) 2. Processes : a. An axon: only one b. Dendrites: one or more TYPES OF NEURONS Based on number of processes 1. Pseudounipolar neurons. 2. Bipolar neurons. 3. Multipolar neurons. TYPES OF NEURONS Based on number of processes 1. Unipolar (Pseudounipolar) neuron (rounded neuron): Has one process only, that divides into two branches; one Dendrite acts as a dendrite and the other as an axon. e.g. Mesencephalic nucleus of Axon trigeminal nerve and dorsal root (spinal) ganglion. TYPES OF NEURONS Based on number of processes 2. Bipolar Neuron (spindle-shaped neuron): Has two processes (one arising from each pole of the cell body). One of them is the dendrite and the other is the axon, e.g. retina & Dendrite olfactory epithelium. TYPES OF NEURONS Based on number of processes 3. Multipolar neuron: Has one axon and multiple dendrites. Types of multipolar neurons: A. Stellate neuron: • The commonest type. • Distributed in most areas of CNS, e.g. anterior horn cells of the spinal cord TYPES OF NEURONS Based on number of processes B. Pyramidal neurons: • Distributed in motor area 4 of the cerebral cortex. C. Pyriform neurons: • Pear-shaped, e.g. Purkinje cells of cerebellar cortex CELL BODY (Perikaryon) Structure of cell body: 1. Nucleus: • Single, usually central, rounded and vesicular with prominent nucleolus. 2. Cytoplasm. CELL BODY (Perikaryon) Cytoplasm: Its main components include: 1. -

Methodic Materials Sensation
Medical Academy named after S.I. Georgievsky of Vernadsky CFU Department of Neurology and Neurosurgery Class 1. Anatomy and Physiology of Sensation. Elements of the Nervous System. Anatomy, physiology, morphology of the Nervous System. Somatosensory System. Pathways. Aids to the examination of the Somatosensory System. Somatosensory Deficits due to Lesions at Specific Sites along the Somatosensory Pathways. Key poin ts: 1. Elements o f the Nervous System. 2. Peripheral Components of the Somatosensory System 3. Posterior Columns. 4. Spinothalamic Tracts. 5. Central Components o f the Somatosensory System 7. Testing for somatosensory deficits. 8. Somatosensory Deficits due to Lesions at Specific Sites along the Somatosensory Pathways Questions for students: Define the following terms: neuron, Nissl substance, axonal transport, Wallerian degeneration, chromatolysis, regeneration of nerve cells, glial cells, pigments and inclusions, nerve fibers, receptor, peripheral nerve, nerve plexus, posterior root, dorsal root ganglion, superficial sensation, deep sensation, posterior columns, spinothalamic pathway, dermatome, conscious proprioception, stereognosis, graphesthesia, 2-point discrimination, anesthesia, hypoesthesia, hyperpathia, allodynia, hyperesthesia, dysesthesia, paresthesia. 1. What are the steps involved in the sensory exam? 2. How is it possible to lose some types of sensations and not others? 3. What sensations are conveyed by the small-diameter sensory nerve fibers in a peripheral nerve? 4. What sensations are conveyed by large-diameter sensory nerve fibers in a peripheral nerve? 5. What sensations are conveyed by the dorsal columns? 6. What sensations are conveyed by the spinothalamic tract? 7. What is tested by double simultaneous stimulation? 8. Where would the lesion be if the patient was able to detect all modalities of sensation but could not recognize an object placed in the right hand? 9. -

Morphological Transformation of Sensory Ganglion Neurons and Satellite Cells
Biomedical Reviews 2000; 11: 39-52. 39 MORPHOLOGICAL TRANSFORMATION OF SENSORY GANGLION NEURONS AND SATELLITE CELLS Seiji Matsuda1, Naoto Kobayashi1, Hiroyuki Wakisaka1, Shouichiro Saito1, Kyouko Saito1, Kyojy Miyawaki1, Katsumi Mominoki2, Kazuhiro Shigemoto3, Shingo Murakami4, and Takashi Fujiwara2 1Department of Anatomy, 2Laboratory Animal Center, 3Department of Hygiene, Ehime University School of Medicine, Shigenobu, Ehime, Japan, 4Department of Otolaryngology, Nagoya City University Medical School, Mizuho, Nagoya, Japan The development of sensory ganglion neurons and satellite cells examined by scanning electron microscopy after removal of the connective tissue is reviewed. Sensory neurons are bipolar at early stages of development and later became pseudounipolar. This maturation event starts earlier but proceeds more slowly in chick than in rat embryos. These may due to the difference in the extent and intimacy of satellite cell investments between these two animal species. The neuronal perikaryal projections are observed by scanning electron microscopy after removal of the connective tissue and satellite cells. The morphometric analysis reveals that perikaryal projections are more numerous on the surface of mature pseudounipolar neurons than on that of premature bipolar neurons; they increase in number as the neuronal cell bodies grow larger. This may support the hypothesis that perikaryal projections are structural devices for increasing the neuron-satellite interface and for improving the efficiency of metabolic exchange between these two cell types.The important role of satellite cells in neuronal maturation is discussed. Biomed Rev 2000; 11: 39-52. INTRODUCTION ganglion neurons, which were first thought to be a technical artifact or an instrument for the attachment between neurons Sensory ganglion neurons undergo a unique transformation and satellite cells, considerably enlarge the perikaryal surface from spindle-shaped bipolar to pseudounipolar cells during (6). -

Unit :3 Nervous System By: Dr
CBCS 3RD SEM (M) PAPER 3026 UNIT :3 NERVOUS SYSTEM BY: DR. LUNA PHUKAN STRUCTURE OF A NEURON A neuron or nerve cell is an electrically excitable cell that communicates with other cells via specialized connections called synapses. It is the main component of nervous tissue in all animals except sponges and placozoa. Plants and fungi do not have nerve cells. The spelling neurone has become uncommon. Neurons are typically classified into three types based on their function. Sensory neurons respond to stimuli such as touch, sound, or light that affect the cells of the sensory organs, and they send signals to the spinal cord or brain. Motor neurons receive signals from the brain and spinal cord to control everything from muscle contractions to glandular output. Interneurons connect neurons to other neurons within the same region of the brain or spinal cord. A group of connected neurons is called a neural circuit. A typical neuron consists of a cell body (soma), dendrites, and a single axon. The soma is usually compact. The axon and dendrites are filaments that extrude from it. Dendrites typically branch profusely and extend a few hundred micrometers from the soma. The axon leaves the soma at a swelling called the axon hillock, and travels for as far as 1 meter in humans or more in other species. It branches but usually maintains a constant diameter. At the farthest tip of the axon's branches are axon terminals, where the neuron can transmit a signal across the synapse to another cell. Neurons may lack dendrites or have no axon. -

NERVOUS SYSTEM: NEURAL TISSUE in Anatomy Today Nervous System Overview • Includes All Neural Tissue in the Body • 2 Divisions 1
Human Anatomy Unit 4 NERVOUS SYSTEM: NEURAL TISSUE In Anatomy Today Nervous System Overview • Includes all neural tissue in the body • 2 divisions 1. Central (CNS) – Brain – Spinal Cord 2. Peripheral (PNS) – Cranial nerves – Spinal nerves – Sensory receptors – Communicates between the CNS and peripheral tissues Nervous System Organization Anatomical Terminology of the Nervous System Central Nervous System Peripheral Nervous System • Control center • Ganglia • Nucleus – Collection of nerve cell bodies – Gray matter – Collection of nerve cell bodies • Spinal nerves • Neural cortex – White matter – Superficial gray matter – Emerge from the spinal cord • Tracts – All mixed nerves – White matter • Cranial nerves • Columns – White matter • Pathways – Emerge from the brain – Sensory – Ascending (sensory) – Motor – Descending (motor) – Mixed Anatomical Terminology of the Nervous System Flow of Information Sensory Motor • Carries sensory information • Carries motor commands from peripheral tissues to from the CNS to peripheral the brain tissues (effectors) • Somatic Sensory • Somatic – All sensory receptors – Controls skeletal muscle throughout the body • Autonomic • Special Sensory – Controls cardiac muscle, – Vision smooth muscle, glands – Hearing – 2 divisions – Equilibrium/balance • Sympathetic – Taste – Fight or flight • Parasympathetic – Smell – Rest and digest Nervous Tissue • 2 distinct types of cells 1. Neurons • Transfer and processing information in the nervous system 2. Neuroglia • Cells that support and protect neurons Neurons Neuroglia -

(Neuron) (Spinal Cord and Brain) Neuroglia (Support) Neuroglia
210 اوم څپرکې عصبي نسج (NERVOUS TISSUE) (impulse) (neuron) (spinal cord and brain) neuroglia (support) neuroglia ٧- ١ شکل دماغ 1012 211 اوم څپرکې عصبي نسج (axons) (NEURON STRUCTURE) cell body Cell body perikaryon Soma ٧-٢ شکل دنيوران جوړښت centrioles granules basal body nissl substance 212 اوم څپرکې عصبي نسج Endoplasmic Reticulum neurotransmitters neurofibrils microtubules microfilaments neurofibrils microtubules substantia (pigmented granules) nerumelanin nigra lipofuscin cell body neuritis Dendrites Axons cell body Dendrites nissle dendrites dendrites granules dendrites cell body 213 اوم څپرکې عصبي نسج axon nissle granuls (axon and dendrites) axon cell body dendrites cell body MAP-2 dendrites dendrites nissl granules axon cell body cell body initial segments axon hillock hillock Schwann cells (sheath) oligodendrocytes Schwann cells Schwann cell ٧- ٤ شکل Schwann cells mesaxon 214 اوم څپرکې عصبي نسج (mesaxon) mesaxon myelin sheath schwann cells myelin sheath neurolimmal sheath neurilemma Schwann cell sheath myelinated axons (myelin sheath) axons Schwann cells myelin sheath myelin 215 اوم څپرکې عصبي نسج node of Ranvier myelin sheath (nodes of Ranvier) (Internode) unmyelinated axons myelin sheath mesaxon Schwann cells Schwann cell body collaterals telodendria boutons terminal boutons of terminaux synapse ganglions synapse 216 اوم څپرکې عصبي نسج dendrites 6,8,3 (nerve fibers) cell body cell bodies cell bodies 120µm cell body 5µm ٧-٨ شکل داکسون او cell body نيوران ارتبات multipolar neurons (bipolar neuron) unipolar neuron bipolar neuron (pseudounipolar -

Neurons: Nerve Cells Are Respo
Exam 3 Anatomy Study Guide Chapter 13: Neural Tissue: 1. How are neurons and neuroglia different? Neurons: nerve cells are responsible for the transfer and processing of information in the nervous system consist of soma, axon, and dendrites Neuroglia: supporting cells, protect the neuron 2. What types of neuroglia are in the CNS and PNS and what are the specific functions of each? PNS: a. Satellite Cells: regulates oxygen and carbon dioxide nutrient, and neurotransmitter levels around neurons in ganglia b. Schwann Cells: responsible for myelination of peripheral axons, and participate in repair process after injury CNS: a. Oligodendrocytes: provide structural framework, myelinate CNS axons b. Astrocytes: maintain blood-brain barrier, provide structural support, regulate ion nutrient c. Microglia: remove cell debris, wastes, and pathogens by phagocytosis d. Ependymal cells: line ventricles (brain), and central canal (spinal cord), assist in producing, circulating and monitoring cerebrospinal fluid 3. What are the components (structure) of a neuron, why cant they reproduce and what are nissl bodies? Dendrites: stimulated by environmental changes or the activities of other cells Cell body: contains the nucleus, mitochondria, ribosomes, and other organelles and inclusions Axon: conducts nerve impulse (action potential) toward synaptic terminals Nissl bodies: create gray matter Neurons cannot reproduce because they lack centrosomes! *4. What are the 5 types of synapses 5. How are neurons classified? Know the 4 structural and 3 functional -

Nervous Tissue
Human Biology/Tissues Nervous Tissue Nervous Tissue: General description: ---Components: nerve cell: neuron Glial cell: neuroglial ---Function: Neurons : receive the stimulation, conduct the nerve impulse. Glial cell: support, protect and insulate, nourish neurons. Neuron: The structural and functional cellular unit. Shape of Neuron: consists of cell body, dendrite and axon. ---cell body(perikaryon or soma): spherical, pyramidal fusiform or stellate in shape. ---dendrite: like branches of tree. ---axon: long thin cord-liked. ( 1 ) Human Biology/Tissues Nervous Tissue Structure of Neuron 1. Cell body: ---cell membrane: unit membrane-irritability, receive stimuli, initiate and conduct nerve impulse membrane proteins-ions channel and receptor ---cell nucleus: large and round, centrally- located, pale stained, with a large, clear nucleolus. ---cytoplasm: perikaryon a. Nissl body: basophilic spot-liked or granule-liked structure, parallelly-arranged rough endoplasmic reticulum and free ribosome. function: synthesis of proteins structural protein enzymes related to the production of neurotransmitters ( 2 ) Human Biology/Tissues Nervous Tissue b. Neurofibril: LM: there are many thread-liked dark brown color structure to form a network. EM: neurofilament microtubule function: supporting involving in intracellular transportation c. other organelle Golgi apparatus mitochondria pigment 2. Dendrite: ---many, short and more branches. ---structure: similar to cell body. ---function: receive the information. 3. Axon: ---thin, less branch. ---structure: no Nissl body, but have microtubule, neurofilament, microfilament, mitochondria, smooth endoplasmic reticulum and vesicle. ( 3 ) Human Biology/Tissues Nervous Tissue axon hillock: the beginning part of axon. axolemma axoplasm ---function: initiate an impulse and conduct the impulse. Classification of Neuron: ---According to number of processes multipolar neuron bipolar neuron pseudounipolar neuron ---According to length of axon Golgi type I neuron: long axon and large. -

The Nervous System 9/24/2009
COGS17 ‐ 01‐The Nervous System 9/24/2009 The Nervous System Mary ET Boyle, Ph.D. Department of Cognitive Science UCSD What’s the big deal? 1 COGS17 ‐ 01‐The Nervous System 9/24/2009 Detect Evaluate Decide Respond Analysis of the nervous system to understand the biological basis for behavior. Modern studies ◦ Neuroanatomists ◦ Neurochemists ◦ Neurophysiologists ◦ Neuropsychologists Neuroscience – term introduced in mid-1960s 2 COGS17 ‐ 01‐The Nervous System 9/24/2009 The brain must know what is happening outside in the environment. •perceive •remember •decide •act sensory neurons motor neurons interneurons Peripheral Nervous System (PNS) Central Nervous System ( CNS) Organization 3 COGS17 ‐ 01‐The Nervous System 9/24/2009 Detects/processes environmental stimuli: Somatic Nervous System Regulates internal organs: Autonomic Nervous System Organization of PNS Detects environmental stimuli: Somatic Nervous System Regulates internal organs: Autonomic Nervous System Decision! Central nervous system 4 COGS17 ‐ 01‐The Nervous System 9/24/2009 Brain Spinal Cord Components of the CNS 5 COGS17 ‐ 01‐The Nervous System 9/24/2009 Anterior Posterior Dl Left Dorsal Right Ventral Anatomical directions 6 COGS17 ‐ 01‐The Nervous System 9/24/2009 Horizontal section— Shows structures viewed from above/below 7 COGS17 ‐ 01‐The Nervous System 9/24/2009 Sagittal section— Divides structures into right and left parts Coronal section (frontal section)— Shows views from the front/back 8 COGS17 ‐ 01‐The Nervous System 9/24/2009 Review of Anatomical Directions Anterior—toward -
Peripheral Nerve Peripheral Nerve (HE) – Cross Section
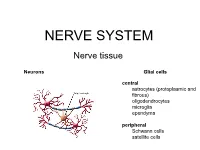
NERVE SYSTEM Nerve tissue Neurons Glial cells central astrocytes (protoplasmic and fibrous) oligodendrocytes microglia ependyma peripheral Schwann cells satellite cells Neurons Glial cells Function in neural network Eferent Sensor Interneurons CNS Astrocytes Sensitive - fibrous (grey matter) Motor - protoplasmic (white matter) Associative Oligodendrocytes Microglia Mediator Ependyme cells Dopaminergic Cholinergic Periphery Serotonergic Schwann cells Aminergic Sattelite cells (amphicytes) GABAergic nitriergic, peptidergic, … Morphology Unipolar Bipolar Multipolar Pseudounipolar Capillaries Axon Golgi type I (long) Golgi type II (short) Classification of neurons According to the number of processes Apolar Unipolar Pseudounipolar Bipolar Multipolar According to the length of axon Golgi I /long axon – 1 m/ Golgi II /short axon - µm/ Apolar – hairy cells in inner ear Unipolar – primary sensory cells /rods, cones/ Bipolar – the 2nd neuron in the retina, ganglion vestibulocochleare Pseudounipolar – T-shaped process. branches of the neurite connecting the ganglion cell with the CNS (central branch) and the periphery (peripheral branch). function as one actively conducting axon, which transmits information from the periphery to the CNS – dorsal root ganglia, some cranial nerves Multipolar - the most frequent – CNS, the autonomic ganglia Spinal multipolar neuroal 10-100m Perikaryon - round nucleus,dominant nucleolus - basophilic cytoplasm – rod-like mitochondria, GA, Nissl substance GER - neurofilaments, neurotubules - lipofuscin