An Independent Locus Upstream of ASIP Controls Variation in the Shade of the Bay Coat Colour in Horses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pferdezucht, -Haltung Und -Fütterung Empfehlungen Für Die Praxis
Sonderheft 353 Special Issue Pferdezucht, -haltung und -fütterung Empfehlungen für die Praxis Wilfried Brade, Ottmar Distl, Harald Sieme und Anette Zeyner (Hrsg.) Landbauforschung vTI Agriculture and Forestry Research Sonderheft 353 Special Issue Preis / Price 10 € LBF_SH_353_U4 9,63 LBF_SH_353_U1 Bibliografische Information der Deutschen Bibliothek Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbiblio- grafie; detaillierte bibliografische Daten sind im Internet über http:// www.d-nb.de/ abrufbar. 2011 Landbauforschung vTI Agriculture and Forestry Research Johann Heinrich von Thünen-Institut Bundesforschungsinstitut für Ländliche Räume, Wald und Fischerei (vTI) Bundesallee 50, D-38116 Braunschweig, Germany Die Verantwortung für die Inhalte liegt bei den jeweiligen Verfassern bzw. Verfasserinnen. [email protected] www.vti.bund.de Preis 10 € ISSN 0376-0723 ISBN 978-3-86576-079-1 LBF_SH 353_U2 9,63 LBF_SH 353_U3 Landbauforschung vTI Agriculture and Forestry Research Sonderheft 353 Special Issue Pferdezucht, -haltung und -fütterung Empfehlungen für die Praxis Wilfried Brade1, Ottmar Distl2, Harald Sieme3 und Anette Zeyner4 (Hrsg.) 1 Stiftung Tierärztl. Hochschule Hannover, zur Zeit: Leibnitz-Institut für Nutztierbiologie (FBN), Wilhelm-Stahl-Allee 2, 18196 Dummerstorf, Email: [email protected] 2 Stiftung Tierärztl. Hochschule Hannover, Institut für Tierzucht und Vererbungsfor- schung, Bünteweg 17 p, 30559 Hannover, Email: [email protected] 3 Stiftung Tierärztl. Hochschule Hannover, Reprod.-med. Einheit der Kliniken, Bünteweg 15, 30559 Hannover, Email: [email protected] 4 Universität Rostock, Professur für Ernährungsphysiologie u. Tierernährung, Justus- von-Liebig-Weg 8, 18059 Rostock, Email: [email protected] W. Brade, O. Distl, H. Sieme, A. Zeyner (Hrsg.), Pferdezucht, -haltung und -fütterung - Empfehlungen für die Praxis Vorwort Lange Zeit befanden sich die meisten Pferde in bäuerlicher Hand. -

CHA TM CHA–THE LEADERS in HORSEMANSHIP SAFETY Changing Lives Through Safe Experiences with Horses •
CHA Certified Horsemanship Association International Conference September 28 – 30, 2018 Colorado State University Equine Sciences CHA TM CHA–THE LEADERS IN HORSEMANSHIP SAFETY Changing Lives Through Safe Experiences with Horses WWW.CHA.HORSE • WWW.CHAINSTRUCTORS.COM CHA CORPORATE OFFICE & STAFF NEW AD RATES 1795 Alysheba Way Ste. 7102 | Lexington, KY 40509 THE INSTRUCTOR 859-259-3399 | [email protected] www.CHA.horse | www.CHAinstructors.com MAGAZINE Christy Landwehr–Chief Executive Officer | 720-857-9550 | [email protected] We print 4,500 issues Terri Weaver–Membership Services Director | 859-259-3399 | [email protected] that are distributed to all Julie Goodnight–International Spokesperson | 719-530-0531 | [email protected] CHA members, CHA individual and equine Executive Committee Board of Directors facilities program members and at our In- Elizabeth Duffy .............................. [email protected] President–Beth Powers ........................ [email protected] Jennifer Eaton ............................... [email protected] ternational and Regional President Elect–Tammi Gainer [email protected] Hayley Eberle ............................. [email protected] Conferences and at all Vice Pres. of Reg. Relations–Anne Brzezicki [email protected] Tara Gamble [email protected] trade shows and events Vice Pres. of New Initiatives–Robert Coleman [email protected] Christine Gillett [email protected] Shellie Hensley -

Essential Genetics for the Horseman
Essential Genetics for the Horseman Whether you are an owner, breeder, rider or other equine enthusiast, there are some essential genetics that can help you to make the most of your journey. This primer is meant to introduce you to the basics of inheritance. Over the last decade, scientific research in equine genetics and the mapping of the equine genome has led to a number of interesting and valuable discoveries. Some of these are directly relevant to the Arabian breed, especially the discoveries relating to disease genes. However, a number of very interesting insights about the entire equine species have been discovered. DNA and Genes The basic starting material for genetics in all species is their DNA. The horse genome (the collection of all the DNA in each cell of the horse) has been completely sequenced, just like in the human. The genome of the horse consists of about 2.7 billion base pairs (the basic unit of DNA, abbreviated by G, A, T, C). This is in comparison to the human genome that has just over 3.0 billion base pairs. When these base pairs are strung together in sets of a few thousand at a time, they provide the ‘blueprint’ for about 20,000 different genes in the horse. About 17,000 of these genes are very similar both in sequence and in function to the corresponding human gene. Chromosomes These genes are physically located on 64 chromosomes in 32 pairs, located in every cell of the horse. Each foal receives a set of 32 chromosomes containing 20,000 genes from its’ dam and also a set from its’ sire. -

Identification of Copy Number Variants in Horses
Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Research Identification of copy number variants in horses Ryan Doan,1 Noah Cohen,2 Jessica Harrington,2 Kylee Veazy,2 Rytis Juras,3 Gus Cothran,3 Molly E. McCue,4 Loren Skow,3 and Scott V. Dindot1,5,6 1Department of Veterinary Pathobiology, 2Department of Large Animal Clinical Sciences, 3Department of Veterinary Integrative Biosciences, College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, Texas 77843, USA; 4Department of Veterinary Population Medicine, University of Minnesota College of Veterinary Medicine, St. Paul, Minnesota 55108, USA; 5Department of Molecular and Cellular Medicine, Texas A&M Health Science Center College of Medicine, College Station, Texas 77843, USA Copy number variants (CNVs) represent a substantial source of genetic variation in mammals. However, the occurrence of CNVs in horses and their subsequent impact on phenotypic variation is unknown. We performed a study to identify CNVs in 16 horses representing 15 distinct breeds (Equus caballus) and an individual gray donkey (Equus asinus) using a whole- exome tiling array and the array comparative genomic hybridization methodology. We identified 2368 CNVs ranging in size from 197 bp to 3.5 Mb. Merging identical CNVs from each animal yielded 775 CNV regions (CNVRs), involving 1707 protein- and RNA-coding genes. The number of CNVs per animal ranged from 55 to 347, with median and mean sizes of CNVs of 5.3 kb and 99.4 kb, respectively. Approximately 6% of the genes investigated were affected by a CNV. Biological process enrichment analysis indicated CNVs primarily affected genes involved in sensory perception, signal transduction, and metabolism. -

Didcot Railway CENTRE
THE COMMUNICATION ORD No. 49 Winter 2018 C Shapland Andrew AWARDS AND ACCOLADES by Graham Langer Tornado in the dark. No. 60163 is seen at Didcot during a night photography session. At the annual Heritage Railway for “reaching out with Tornado to new film. Secondly we scooped the John Association awards ceremony held at the and wider audiences” in recognition Coiley Locomotive Engineering award for Burlington Arcade Hotel in Birmingham of the locomotive’s adventures in the work associated with the 100mph on 10th February, the Trust was 2017, initially on the ‘Plandampf’ series run. Trustees and representatives of DB honoured to be awarded not one but on the Settle & Carlisle railway, then Cargo, Ricardo Rail, Resonate, Darlington two national prizes. Firstly we received the 100mph run and its associated Borough Council and the Royal Navy the Steam Railway Magazine Award, television coverage and finally in her were among the Trust party who ➤ presented by editor Nick Brodrick, appearance in the PADDINGTON 2 attended the event. TCC 1 Gwynn Jones CONTENTS EDItorIAL by Graham Langer PAGE 1-2 Mandy Gran Even while Tornado Awards and Accolades up his own company Paul was Head of PAGE 3 was safely tucked Procurement for Northern Rail and Editorial up at Locomotive previously Head of Property for Arriva Tornado helps Blue Peter Maintenance Services Trains Northern. t PAGE 4 in Loughborough Daniela Filova,´ from Pardubice in the Tim Godfrey – an obituary for winter overhaul, Czech Republic, joined the Trust as Richard Hardy – an obituary she continued to Assistant Mechanical Engineer to David PAGE 5 generate headlines Elliott. -

The Last Populations of the Critically Endangered Onager Equus Hemionus Onager in Iran: Urgent Requirements for Protection and Study
Oryx Vol 37 No 4 October 2003 Short Communication The last populations of the Critically Endangered onager Equus hemionus onager in Iran: urgent requirements for protection and study Laurent Tatin, Bijan F. Darreh-Shoori, Christophe Tourenq, David Tatin and Bijan Azmayesh Abstract The onager Equus hemionus onager, a wild ass for domestic use, and land conversion have been identi- endemic to Iran, is categorized as Critically Endangered fied as the main threats to the two remaining onager on the IUCN Red List. Its biology and conservation populations. In addition, geographical isolation could requirements are poorly documented. We report our cause the loss of genetic variability in these two relatively observations, made in 1997 and 2000, on the behaviour small populations, and also makes them more susceptible and ecology of the two remaining populations, located to the potential eCects of stochastic events such as drought in the Touran Protected Area and the Bahram-e-Goor or disease. Public awareness, appropriate protection, and Reserve. Recent population counts by the Department of scientific studies must be urgently supported by both Environment of Iran (471 in the Protected Area and 96 national and international organizations in order to pre- in the Reserve) are markedly lower than the estimate of vent the extinction of these two apparently dwindling 600–770 made in the 1970s in the Touran Protected Area. populations of onager. We observed social interactions between stallions and mares outside the breeding season that contrasts with Keywords Ass, behaviour, conservation status, Equus the known social structure of this subspecies. Poaching, hemionus, Iran, onager. competition with domestic animals, removal of shrubs The Asiatic wild ass Equus hemionus is one of seven ass Equus h. -

Biodiversity of Arabian Horses in Syria
Biodiversity of Arabian horses in Syria Dissertation zur Erlangung des akademischen Grades Doctor rerum agriculturarum (Dr. rer. agr.) eingereicht an der Lebenswissenschaftlichen Fakultät der Humboldt Universität zu Berlin von M.Sc. Saria Almarzook Präsidentin der Humboldt-Universität zu Berlin Prof. Dr. Sabine Kunst Dekan der Humboldt-Universität zu Berlin Prof. Dr. Bernhard Grimm Gutachterin/Gutachter Prof. Dr. Gudrun Brockmann Prof. Dr. Dirk Hinrichs Prof. Dr. Armin Schmitt Tag der mündlichen Prüfung: 17. September 2018 Dedication This research is dedicated to my homeland …Syria Contents Zusammenfassung ................................................................................................................... I Summary ............................................................................................................................... VI List of publications and presentations .................................................................................. XII List of abbreviations ............................................................................................................ XIII List of figures ....................................................................................................................... XIV List of tables ......................................................................................................................... XV 1. General introduction and literature review ..................................................................... 1 1.1. Domestication and classification -

Unraveling the Evolutionary History of Orangutans (Genus: Pongo)- the Impact of Environmental Processes and the Genomic Basis of Adaptation
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2015 Unraveling the evolutionary history of Orangutans (genus: Pongo)- the impact of environmental processes and the genomic basis of adaptation Mattle-Greminger, Maja Patricia Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-121397 Dissertation Published Version Originally published at: Mattle-Greminger, Maja Patricia. Unraveling the evolutionary history of Orangutans (genus: Pongo)- the impact of environmental processes and the genomic basis of adaptation. 2015, University of Zurich, Faculty of Science. Unraveling the Evolutionary History of Orangutans (genus: Pongo) — The Impact of Environmental Processes and the Genomic Basis of Adaptation Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch‐naturwissenschaftlichen Fakultät der Universität Zürich von Maja Patricia Mattle‐Greminger von Richterswil (ZH) Promotionskomitee Prof. Dr. Carel van Schaik (Vorsitz) PD Dr. Michael Krützen (Leitung der Dissertation) Dr. Maria Anisimova Zürich, 2015 To my family Table of Contents Table of Contents ........................................................................................................ 1 Summary ..................................................................................................................... 3 Zusammenfassung ..................................................................................................... -
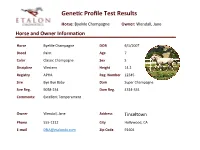
Gene C Profile Test Results
GeneGc Profile Test Results Horse: ByeMe Champagne Owner: Wendall, Jane Horse and Owner Informaon Horse ByeMe Champagne DOB 6/1/2007 Breed Paint Age 7 Color Classic Champagne Sex S Discipline Western Height 14.2 Registry APHA Reg. Number 12345 Sire Bye Bye Baby Dam Super Champagne Sire Reg. 9058-234 Dam Reg. 4314-334 Comments: Excellent Temperament Owner Wendall, Jane Address Tinseltown Phone 555-1212 City Hollywood, CA E-mail [email protected] Zip Code 91604 GeneGc Profile Test Results Horse: ByeMe Champagne Owner: Wendall, Jane Results Summary Coat Color : ByeMe Champagne has one Black allele and one Red allele making the Base coat appear Black. Also detected were single Champagne and Cream alleles; likely resulUng in a rare Champagne Cream color. One copy of the Frame Overo allele is also detected, indicang underlying white patches (hidden By CH). As a result of single gene copies in each of the following, he has a 50% chance of passing Black or Red, Cream and/or Champagne, AgouU and/or Frame Overo alleles to his offspring. Allele Summary: Aa, Ee, Ch, Cr, LWO/n Traits: ByeMe Champagne is a not a carrier of any known recessive disease genes. CauUon is recommended however, as any mare Bred to him should Be Frame Overo negave as to avoid a 25% chance of foal death (+/+ LWO results in a lethal condiUon at Birth). He may also throw Gaited foals when Bred to Gaited (+) mares. Notes: Please note that your analysis is ongoing and may include some regions marked with an asterisk denoUng the following: * Discovery – This gene detecUon is in the -

Reintroduction of Przewalski’S Horse (Equus Ferus Przewalskii)
Biological Conservation 177 (2014) 142–147 Contents lists available at ScienceDirect Biological Conservation journal homepage: www.elsevier.com/locate/biocon Short communication Reintroduction of Przewalski’s horse (Equus ferus przewalskii) in Xinjiang, China: The status and experience ⇑ Canjun Xia a, Jie Cao b, Hefan Zhang b, Xingyi Gao a, Weikang Yang a, , David Blank a a Key Laboratory of Biogeography and Bioresources in Arid Land, Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, South Beijing Road 818, Urumqi 830011, Xinjiang, China b Wild Horse Breeding Centre, Jimsar 831700, Xinjiang, China article info abstract Article history: Przewalski’s horse reintroductions to Xinjiang, China were initiated in 1985. Here, we present the first Received 13 January 2014 data on population development and current problems of the Przewalski’s horse in both captive and Received in revised form 20 June 2014 released populations in Xinjiang. From 1985 to 2005, a total of 24 captive Przewalski’s horses (14 males Accepted 23 June 2014 and 10 females) were brought from western zoos to the Jimsar Wild Horse Breeding Center (WHBC) in Xinjiang. In 1988, the first foal was born. Since then, a total of 285 foals have been born and the number of animals in the captive population continues to increase. In August 2001, the first group of horses was Keywords: released into semi-wild conditions in the Kalamaili Nature Reserve (KNR). Released horses were allowed Asian wild horse to range freely from spring to fall, but were driven into a winter coral to allow for supplemental feeding Reintroduction Equus ferus przewalskii and to increase winter survival, and to reduce competition with domestic horses from local herdsmen China who use the KNR as winter pasture. -

Late-Pleistocene Horse (Equus Sp.) from the Wilson-Leonard Archaeological Site, Central Texas
CURRENT RESEARCH IN THE PLEISTOCENE Vol. 19, 2002 Paleoenvironmental: Vertebrates Late-Pleistocene Horse (Equus sp.) from the Wilson-Leonard Archaeological Site, Central Texas Barry W. Baker, Michael B. Collins and C. Britt Bousman The Wilson-Leonard site (41WM235) near Austin represents one of the best preserved and dated, long-term archaeological sequences in the Southern Plains. Occupations are pre-Clovis through late-Prehistoric in age (Collins 1998; Collins et al. 1993). The site is located on Brushy Creek in Williamson County, on the eastern edge of the Edwards Plateau along an ecotone with the Black Prairie. The Wilson-Leonard site has received wide attention for the recovery a late-Paleoindian human female skeleton (Wilson Component), as well as for the generalized human diet inferred for the late-Pleistocene / early-Holocene transition (Bousman 1998). Extensive subsistence and environmental data have been reported, including a well preserved vertebrate faunal assemblage (Baker 1994, 1998a, 1998b, 1998c; Balinsky 1997, 1998; Decker 1998, and Winkler 1990). Within the faunal assemblage is a single horse bone (Equus sp.). Following the nomenclature of Driesch (1976:91) and Peters (1987), the bone is a complete left central tarsal (os tarsi centrale [navicular]. Archaeological provenience is as follows: Excavation square E28/S78; Level 39A&B; Stratigraphic Unit Isi/Icl. The central tarsal is not burned, and no cut marks or other forms of potential cultural modification were observed. This bone was recovered from an area of the site referred to as the Bone Bed Component. The Texas Department of Transportation (TxDOT) recovered the bone during 1982-1984 excavations at the site. -

The Base Colors: Black and Chestnut the Tail, Called “Foal Fringes.”The Lower Legs Can Be So Pale That It Is Let’S Begin with the Base Colors
Foal Color 4.08 3/20/08 2:18 PM Page 44 he safe arrival of a newborn foal is cause for celebration. months the sun bleaches the foal’s birth coat, altering its appear- After checking to make sure all is well with the mare and ance even more. Other environmental issues, such as type and her new addition, the questions start to fly. What gender quality of feed, also can have a profound effect on color. And as we is it? Which traits did the foal get from each parent? And shall see, some colors do change drastically in appearance with Twhat color is it, anyway? Many times this question is not easily age, such as gray and the roany type of sabino. Finally, when the answered unless the breeder has seen many foals, of many colors, foal shed occurs, the new color coming in often looks dramatical- throughout many foaling seasons. In the landmark 1939 movie, ly dark. Is it any wonder that so many foals are registered an incor- “The Wizard of Oz,” MGM used gelatin to dye the “Horse of a rect—and sometimes genetically impossible—color each year? Different Color,” but Mother Nature does a darn good job of cre- So how do you identify your foal’s color? First, let’s keep some ating the same spectacular special effects on her foals! basic rules of genetics in mind. Two chestnuts will only produce The foal’s color from birth to the foal shed (which generally chestnut; horses of the cream, dun, and silver dilutions must have occurs between three and four months of age) can change due to had at least one parent with that particular dilution themselves; many factors, prompting some breeders to describe their foal as and grays must always have one gray parent.