Comparative and Phylogenomic Studies on the Mitochondrial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Insetos Do Brasil
COSTA LIMA INSETOS DO BRASIL 2.º TOMO HEMÍPTEROS ESCOLA NACIONAL DE AGRONOMIA SÉRIE DIDÁTICA N.º 3 - 1940 INSETOS DO BRASIL 2.º TOMO HEMÍPTEROS A. DA COSTA LIMA Professor Catedrático de Entomologia Agrícola da Escola Nacional de Agronomia Ex-Chefe de Laboratório do Instituto Oswaldo Cruz INSETOS DO BRASIL 2.º TOMO CAPÍTULO XXII HEMÍPTEROS ESCOLA NACIONAL DE AGRONOMIA SÉRIE DIDÁTICA N.º 3 - 1940 CONTEUDO CAPÍTULO XXII PÁGINA Ordem HEMÍPTERA ................................................................................................................................................ 3 Superfamília SCUTELLEROIDEA ............................................................................................................ 42 Superfamília COREOIDEA ............................................................................................................................... 79 Super família LYGAEOIDEA ................................................................................................................................. 97 Superfamília THAUMASTOTHERIOIDEA ............................................................................................... 124 Superfamília ARADOIDEA ................................................................................................................................... 125 Superfamília TINGITOIDEA .................................................................................................................................... 132 Superfamília REDUVIOIDEA ........................................................................................................................... -

(Pentatomidae) DISSERTATION Presented
Genome Evolution During Development of Symbiosis in Extracellular Mutualists of Stink Bugs (Pentatomidae) DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Alejandro Otero-Bravo Graduate Program in Evolution, Ecology and Organismal Biology The Ohio State University 2020 Dissertation Committee: Zakee L. Sabree, Advisor Rachelle Adams Norman Johnson Laura Kubatko Copyrighted by Alejandro Otero-Bravo 2020 Abstract Nutritional symbioses between bacteria and insects are prevalent, diverse, and have allowed insects to expand their feeding strategies and niches. It has been well characterized that long-term insect-bacterial mutualisms cause genome reduction resulting in extremely small genomes, some even approaching sizes more similar to organelles than bacteria. While several symbioses have been described, each provides a limited view of a single or few stages of the process of reduction and the minority of these are of extracellular symbionts. This dissertation aims to address the knowledge gap in the genome evolution of extracellular insect symbionts using the stink bug – Pantoea system. Specifically, how do these symbionts genomes evolve and differ from their free- living or intracellular counterparts? In the introduction, we review the literature on extracellular symbionts of stink bugs and explore the characteristics of this system that make it valuable for the study of symbiosis. We find that stink bug symbiont genomes are very valuable for the study of genome evolution due not only to their biphasic lifestyle, but also to the degree of coevolution with their hosts. i In Chapter 1 we investigate one of the traits associated with genome reduction, high mutation rates, for Candidatus ‘Pantoea carbekii’ the symbiont of the economically important pest insect Halyomorpha halys, the brown marmorated stink bug, and evaluate its potential for elucidating host distribution, an analysis which has been successfully used with other intracellular symbionts. -
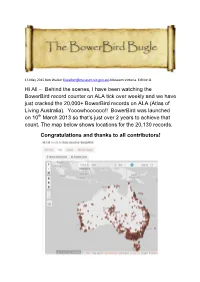
Behind the Scenes, I Have Been Watching the Bowerbird Record
15 May 2015 Ken Walker ([email protected]) Museum Victoria. Edition 8. Hi All – Behind the scenes, I have been watching the BowerBird record counter on ALA tick over weekly and we have just cracked the 20,000+ BowerBird records on ALA (Atlas of Living Australia). Yooowhoooooo!! BowerBird was launched on 10th March 2013 so that’s just over 2 years to achieve that count. The map below shows locations for the 20,130 records. Congratulations and thanks to all contributors! From a purely funding analysis point of view, the initial ALA grant to develop BowerBird was $350,000 which at 20,130 records is currently returning per record at a cost of $17.38 and that per record cost will only get lower as more BowerBird records are added and uploaded. For some species on ALA, BowerBird provides the only distributional species data points: For other species on ALA, BowerBird provides the only species image for species on ALA: From a Biosecurity point of view of tracking exotic species, BowerBird has supplied all records post 2007 for the exotic South African Carder bee. All of the blue dots represent BowerBird records. Other than the great biodiversity results that BowerBird delivers through ALA, there are a myriad of other intangible benefits that come from the BowerBird website. Intangibles such as the member’s conversations, identification discussions, the friendships, the biological statements and the generated innate knowledge. One particular “intangible”, I would like to tell you about here. The PBCRC (Plant Biosecurity, Cooperative Research Centre) has a theme called “Building Resilience through Remote Indigenous Engagement”. -

(Pueraria Montana Var. Lobata Willd) in North Carolina
ABSTRACT THORNTON, MELISSA ROSE. Arthopod Fauna Associated With Kudzu (Pueraria montana var. lobata Willd) In North Carolina. (Under the direction of David Orr.) The purpose of this research was to obtain background information to aid the implementation of a biological control program against the weed, kudzu (Pueraria montana var. lobata Willd). This research had several specific objectives that examined: 1) potential insect pollinators and seed production of kudzu in NC; 2) phytophagous insects and insect herbivory of kudzu foliage, seeds, vines and roots in NC; 3) abundance and diversity of foliar, vine, and root feeding insect communities on kudzu in comparison with those found on soybeans, the closest North American relative of kudzu in the United States. Kudzu is pollinated by native and naturalized insects in NC, in a pattern that varies by flower apparency rather than density. Arthropod herbivory by native generalists almost eliminated kudzu seed viability, while a naturalized Asian specialist consumed a nominal proportion of seeds. These data indicate that seed feeding arthropods would be poor candidates for importation biological control. Kudzu and soybeans shared the same foliar feeding insect communities and levels of defoliation, suggesting that foliage feeders are also poor choices for importation. No kudzu vine or root feeding insects or damage were found during the two years of this study, suggesting that future importation biological control research should focus on such feeders from Asia. ARTHROPOD FAUNA ASSOCIATED WITH KUDZU (PUERARIA MONTANA VAR. LOBATA WILLD) IN NORTH CAROLINA by MELISSA ROSE THORNTON A thesis submitted to the Graduate Faculty of North Carolina State University in partial fulfillment of the requirements for the Degree of Master of Science DEPARTMENT OF ENTOMOLOGY Raleigh 2004 APPROVED BY: ______________________________ ______________________________ Dr. -

And Phylogenetic Implications
G C A T T A C G G C A T genes Article The Complete Mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and Phylogenetic Implications Qi-Lin Zhang 1,2 , Run-Qiu Feng 1, Min Li 1, Zhong-Long Guo 1 , Li-Jun Zhang 1, Fang-Zhen Luo 1, Ya Cao 1 and Ming-Long Yuan 1,* 1 State Key Laboratory of Grassland Agro-ecosystems; Key Laboratory of Grassland Livestock Industry Innovation, Ministry of Agriculture and Rural Affairs; Engineering Research Center of Grassland Industry, Ministry of Education; College of Pastoral Agriculture Science and Technology, Lanzhou University, Lanzhou 730000, China; [email protected] (Q.-L.Z.); [email protected] (R.-Q.F.); [email protected] (M.L.); [email protected] (Z.-L.G.); [email protected] (L.-J.Z.); [email protected] (F.-Z.L.); [email protected] (Y.C.) 2 Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming 650500, China * Correspondence: [email protected] Received: 29 August 2019; Accepted: 15 October 2019; Published: 18 October 2019 Abstract: We determined the complete mitogenome of Pyrrhocoris tibialis (Hemiptera: Heteroptera: Pyrrhocoridae) to better understand the diversity and phylogeny within Pentatomomorpha, which is the second largest infra-order of Heteroptera. Gene content, gene arrangement, nucleotide composition, codon usage, ribosomal RNA (rRNA) structures, and sequences of the mitochondrial transcription termination factor were well conserved in Pyrrhocoroidea. Different protein-coding genes have been subject to different evolutionary rates correlated with the G + C content. The size of control regions (CRs) was highly variable among mitogenomes of three sequenced Pyrrhocoroidea species, with the P. -

Economic Injury Level of the Neotropical Brown Stink Bug Euschistus Heros (F.) on Cotton Plants
Neotrop Entomol (2017) 46:324–335 DOI 10.1007/s13744-016-0454-2 PEST MANAGEMENT Economic Injury Level of the Neotropical Brown Stink Bug Euschistus heros (F.) on Cotton Plants 1 1 2 3 MF SORIA ,PEDEGRANDE ,ARPANIZZI ,MDTOEWS 1Lab of Applied Entomology and Biotechnology, Graduate Program in Agronomy, School of Agricultural Sciences, Federal Univ of Grande Dourados, Dourados, MS, Brasil 2Lab of Entomology, Embrapa National Wheat Research Center, Passo Fundo, RS, Brasil 3Dept of Entomology, Univ of Georgia, Tifton, GA, USA Keywords Abstract Pentatomidae, sucking pest, attack, injury, In Brazil, the Neotropical brown stink bug, Euschistus heros (F.) damage, treatment threshold (Hemiptera: Pentatomidae), commonly disperses from soybeans to Correspondence cotton fields. The establishment of an economic treatment threshold MF Soria, Lab of Applied Entomology and for this pest on cotton crops is required. Infestation levels of adults of Biotechnology, Graduate Program in Agronomy, School of Agricultural Sciences, E. heros were evaluated on cotton plants at preflowering, early Federal Univ of Grande Dourados, flowering, boll filling, and full maturity by assessing external and Dourados, MS, Brasil; miguelagro@gmail. internal symptoms of injury on bolls, seed cotton/lint production, com and fiber quality parameters. A completely randomized experiment Edited by Jorge B Torres – UFRPE was designed to infest cotton plants in a greenhouse with 0, 2, 4, 6, and 8 bugs/plant, except at the full-maturity stage in which only Received 13 March 2016 and accepted 13 October 2016 infestation with 8 bugs/plant and uninfested plants were evaluated. Published online: 14 November 2016 Results indicated that the preflowering, early-flowering, and full- maturity stages were not affected by E. -

A New Family of Coreoidea from the Lower Cretaceous Lebanese Amber (Hemiptera: Pentatomomorpha)
P O L I S H JOUR NAL OF ENTOMOLO G Y POLSKIE PISMO ENTOMOL OGICZ N E VOL. 80: 627-644 Gdynia 31 December 2011 DOI: 10.2478/v10200-011-0049-5 A new family of Coreoidea from the Lower Cretaceous Lebanese Amber (Hemiptera: Pentatomomorpha) DANY AZAR1, ANDRÉ NEL2, MICHAEL S. ENGEL3, 4, ROMAIN GARROUSTE2, ARMAND MATOCQ2 1Lebanese University, Faculty of Sciences II, Department of Natural Sciences, PO box 26110217, Fanar – Matn, Lebanon, e-mail: [email protected]; 2CNRS UMR 7205, CP 50, Entomologie, Muséum National d'Histoire Naturelle, 45 rue Buffon, F-75005 Paris, France, e-mails: [email protected], [email protected], [email protected]; 3Division of Entomology (Paleoentomology), Natural History Museum, University of Kansas, Lawrence, KS 66049-2811, USA, e-mail: [email protected]; 4Department of Ecology & Evolutionary Biology, 1501 Crestline Drive – Suite 140, University of Kansas, Lawrence, KS 66049-2811, USA ABSTRACT. A new genus and species, Yuripopovina magnifica, belonging to a new coreoid family, Yuripopovinidae (Hemiptera: Pentatomomorpha), is described and illustrated from the Lower Cretaceous amber of Lebanon. The species represents the first definitive Mesozoic record for the Coreoidea. A cladistic analysis of Coreoidea, including the new family, is undertaken. KEY WORDS: Pentatomomorpha, Coreoidea, Yuripopovinidae, fam. n., gen. n., sp. n., Lebanon, phylogeny. INTRODUCTION The Pentatomomorpha with its 14 000 known living species (WEIRAUCH & SCHUH 2011) is the second largest of the seven heteropteran infraorders (SCHAEFER 1993, ŠTYS & KERZHNER 1975) (Enicocephalomorpha, Dipsocoromorpha, Gerromorpha, Nepomorpha, Leptodomorpha, Cimicomorpha, and Pentatomorpha). Most authors recognize five superfamilies within Pentatomomorpha, but there remains controversy regarding the 628 Polish Journal of Entomology 80 (4) composition of these superfamilies (SCHAEFER 1993, ŠTYS 1961). -

An Overview of Flat Bug Genera (Hemiptera, Aradidae)
ZOBODAT - www.zobodat.at Zoologisch-Botanische Datenbank/Zoological-Botanical Database Digitale Literatur/Digital Literature Zeitschrift/Journal: Denisia Jahr/Year: 2006 Band/Volume: 0019 Autor(en)/Author(s): Lariviere Marie C., Larochelle Andre Artikel/Article: An overview of flat bug genera (Hemiptera, Aradidae) from New Zealand, with considerations on faunal diversification and affinities 181-214 © Biologiezentrum Linz/Austria; download unter www.biologiezentrum.at An overview of flat bug genera (Hemiptera, Aradidae) from New Zealand, with considerations on faunal diversification and affinities1 M .-C . L ARIVIÈRE & A. L AROCHELLE Abstract: Nineteen genera and thirty-nine species of Aradidae have been described from New Zealand, most of which are endemic (12 genera, 38 species). An overview of all genera and an identification key to subfamilies, tribes, and genera are presented for the first time. Species included in each genus are list- ed for New Zealand. Concise generic descriptions, illustrations emphasizing key diagnostic features, colour photographs representing each genus, an overview of the most relevant literature, and notes on distribution are also given. The biology and diversification of New Zealand aradids, and their affinities with neighbouring faunas are briefly discussed. Key words: Aradidae, biogeography, Hemiptera, New Zealand, taxonomy. Introduction forests, and using their stylets to extract liq- uids from fungal hyphae associated with de- The Aradidae, also commonly referred caying wood. Many ground-dwelling species to as flat bugs or bark bugs, form a large fam- of rainforest environments are wingless – a ily of Heteroptera containing over 1,800 condition thought to have evolved several species and 210 genera worldwide (SCHUH times in the phylogeographic history of the & SLATER 1995). -

Insect Classification Standards 2020
RECOMMENDED INSECT CLASSIFICATION FOR UGA ENTOMOLOGY CLASSES (2020) In an effort to standardize the hexapod classification systems being taught to our students by our faculty in multiple courses across three UGA campuses, I recommend that the Entomology Department adopts the basic system presented in the following textbook: Triplehorn, C.A. and N.F. Johnson. 2005. Borror and DeLong’s Introduction to the Study of Insects. 7th ed. Thomson Brooks/Cole, Belmont CA, 864 pp. This book was chosen for a variety of reasons. It is widely used in the U.S. as the textbook for Insect Taxonomy classes, including our class at UGA. It focuses on North American taxa. The authors were cautious, presenting changes only after they have been widely accepted by the taxonomic community. Below is an annotated summary of the T&J (2005) classification. Some of the more familiar taxa above the ordinal level are given in caps. Some of the more important and familiar suborders and families are indented and listed beneath each order. Note that this is neither an exhaustive nor representative list of suborders and families. It was provided simply to clarify which taxa are impacted by some of more important classification changes. Please consult T&J (2005) for information about taxa that are not listed below. Unfortunately, T&J (2005) is now badly outdated with respect to some significant classification changes. Therefore, in the classification standard provided below, some well corroborated and broadly accepted updates have been made to their classification scheme. Feel free to contact me if you have any questions about this classification. -

Integrating Cultural Tactics Into the Management of Bark Beetle and Reforestation Pests1
DA United States US Department of Proceedings --z:;;-;;; Agriculture Forest Service Integrating Cultural Tactics into Northeastern Forest Experiment Station the Management of Bark Beetle General Technical Report NE-236 and Reforestation Pests Edited by: Forest Health Technology Enterprise Team J.C. Gregoire A.M. Liebhold F.M. Stephen K.R. Day S.M.Salom Vallombrosa, Italy September 1-3, 1996 Most of the papers in this publication were submitted electronically and were edited to achieve a uniform format and type face. Each contributor is responsible for the accuracy and content of his or her own paper. Statements of the contributors from outside the U.S. Department of Agriculture may not necessarily reflect the policy of the Department. Some participants did not submit papers so they have not been included. The use of trade, firm, or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the U.S. Department of Agriculture or the Forest Service of any product or service to the exclusion of others that may be suitable. Remarks about pesticides appear in some technical papers contained in these proceedings. Publication of these statements does not constitute endorsement or recommendation of them by the conference sponsors, nor does it imply that uses discussed have been registered. Use of most pesticides is regulated by State and Federal Law. Applicable regulations must be obtained from the appropriate regulatory agencies. CAUTION: Pesticides can be injurious to humans, domestic animals, desirable plants, and fish and other wildlife - if they are not handled and applied properly. -

Hemiptera, Prosorrhyncha) with Special Reference to the Pregenital Abdominal Structure1
© Biologiezentrum Linz/Austria; download unter www.biologiezentrum.at Justification for the Aradimorpha as an infraorder of the suborder Heteroptera (Hemiptera, Prosorrhyncha) with Special Reference to the Pregenital Abdominal Structure1 M.H. SWEET Abstract: Aradomorpha SWEET 1996 is replaced with Aradimorpha because of homonymy with Arado- morpha CHAMPION 1899, a genus of Reduviidae. The Aradimorpha differ from the Pentatomomorpha s.s. and the Leptopodomorpha in having a plesiomorphic connexivum of dorsal epipleurites and ventral hy- popleurites rather than having the connexivum turned over so that the hypopleurites are dorsalized and the epipleurites folded into the abdomen. In most Aradimorpha, in both males and females, sterna 3 to 7 are free with intersegmental conjunctiva; terga 1-2 and 3 to 6 are united, but all epipleurites are free. In the Pentatomomorpha at least abdominal sterna 2 to 4 in females and sterna 2 to 5 in males are uni- ted or fused without conjunctiva. In some aradids the hypopleurites are united or fused with the sterna, but hypopleurite 2 is usually free. Sternum 2 is sometimes united to fused with sternum 1 and the meta- sternum. The abdominal spiracles in the Aradimorpha are ventral on the hypopleurites, although some- times very lateral in position on the hypopleurites, with the exception of the Chinamyersiini in which spiracles 4, 5 and 6 are dorsal on the epipleurites in Chinamyersia, and 5 and 6 dorsal in Gnostocoris, whi- le in the Tretocorini (Tretocoris and Kumaressa) spiracle 2 seems dorsal but is actually very lateral on the hypopleurite. In the Termitaphididae, epipleurites and hypopleurites are distinct, forming mobile lateral abdominal lobes. -

Mitochondrial Markers for Identification and Phylogenetic Studies in Insects – a Review
DOI: 10.2478/dna-2014-0001 DNA Barcodes 2014; Volume 2: 1–9 Research Article Open Access Surajit De Mandal, Liansangmawii Chhakchhuak, Guruswami Gurusubramanian, Nachimuthu Senthil Kumar* Mitochondrial markers for identification and phylogenetic studies in insects – A Review Abstract: Similar morphology and high genetic diversity stranded molecule with a range of 14,503 bp (Rhopalomyia poses problems in phylogenetic studies of insects. To pomum) to 19,517 bp (Drosophila melanogaster) in size solve these problems, mitochondrial based markers have [7]. It consists of 37 genes encoding the large and small been adopted and are increasingly used as molecular subunit ribosomal RNAs, 22 transfer RNAs (trnI, trnQ, markers for phylogenetic studies. Varied markers have trnM, trnW, trnC, trnY, trnL1, trnK, trnD, trnG, trnA, trnR, been used for different species of insects, viz., markers for trnN, trnS1, trnE, trnF, trnH, trnT, trnP, trnS2, trnL2, trnV) 16S r RNA, 12S r RNA, ND (1-6 genes), ATPase and control necessary to translate the protein-coding genes and regions. Among which protein coding gene, CO-1 is found 13 protein coding genes that are all components of the to be best because of its advantage over others whereas, oxidative phosphorylation process (Figure 1). The insect AT rich region of mitochondrial DNA is the least used mt genome also consists of a regulatory element known as marker. A recent advanced technology in phylogenetic AT rich region which plays important role in initiation of analysis; namely mitogenomics have greatly improved this transcription and replication [8,9]. research field. This short review attempted to summarize The arrangement of major genes in the mitochondrial recent studies on the application of vari ous mitochondrial genome is highly conserved across animal phyla.