Source Apportionment of Ambient Mercury at Flin Flon, Manitoba
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Valid Operating Permits
Valid Petroleum Storage Permits (as of September 15, 2021) Permit Type of Business Name City/Municipality Region Number Facility 20525 WOODLANDS SHELL UST Woodlands Interlake 20532 TRAPPERS DOMO UST Alexander Eastern 55141 TRAPPERS DOMO AST Alexander Eastern 20534 LE DEPANNEUR UST La Broquerie Eastern 63370 LE DEPANNEUR AST La Broquerie Eastern 20539 ESSO - THE PAS UST The Pas Northwest 20540 VALLEYVIEW CO-OP - VIRDEN UST Virden Western 20542 VALLEYVIEW CO-OP - VIRDEN AST Virden Western 20545 RAMERS CARWASH AND GAS UST Beausejour Eastern 20547 CLEARVIEW CO-OP - LA BROQUERIE GAS BAR UST La Broquerie Red River 20551 FEHRWAY FEEDS AST Ridgeville Red River 20554 DOAK'S PETROLEUM - The Pas AST Gillam Northeast 20556 NINETTE GAS SERVICE UST Ninette Western 20561 RW CONSUMER PRODUCTS AST Winnipeg Red River 20562 BORLAND CONSTRUCTION INC AST Winnipeg Red River 29143 BORLAND CONSTRUCTION INC AST Winnipeg Red River 42388 BORLAND CONSTRUCTION INC JST Winnipeg Red River 42390 BORLAND CONSTRUCTION INC JST Winnipeg Red River 20563 MISERICORDIA HEALTH CENTRE AST Winnipeg Red River 20564 SUN VALLEY CO-OP - 179 CARON ST UST St. Jean Baptiste Red River 20566 BOUNDARY CONSUMERS CO-OP - DELORAINE AST Deloraine Western 20570 LUNDAR CHICKEN CHEF & ESSO UST Lundar Interlake 20571 HIGHWAY 17 SERVICE UST Armstrong Interlake 20573 HILL-TOP GROCETERIA & GAS UST Elphinstone Western 20584 VIKING LODGE AST Cranberry Portage Northwest 20589 CITY OF BRANDON AST Brandon Western 1 Valid Petroleum Storage Permits (as of September 15, 2021) Permit Type of Business Name City/Municipality -

Manitoba Regional Health Authority (RHA) DISTRICTS MCHP Area Definitions for the Period 2002 to 2012
Manitoba Regional Health Authority (RHA) DISTRICTS MCHP Area Definitions for the period 2002 to 2012 The following list identifies the RHAs and RHA Districts in Manitoba between the period 2002 and 2012. The 11 RHAs are listed using major headings with numbers and include the MCHP - Manitoba Health codes that identify them. RHA Districts are listed under the RHA heading and include the Municipal codes that identify them. Changes / modifications to these definitions and the use of postal codes in definitions are noted where relevant. 1. CENTRAL (A - 40) Note: In the fall of 2002, Central changed their districts, going from 8 to 9 districts. The changes are noted below, beside the appropriate district area. Seven Regions (A1S) (* 2002 changed code from A8 to A1S *) '063' - Lakeview RM '166' - Westbourne RM '167' - Gladstone Town '206' - Alonsa RM 'A18' - Sandy Bay FN Cartier/SFX (A1C) (* 2002 changed name from MacDonald/Cartier, and code from A4 to A1C *) '021' - Cartier RM '321' - Headingley RM '127' - St. Francois Xavier RM Portage (A1P) (* 2002 changed code from A7 to A1P *) '090' - Macgregor Village '089' - North Norfolk RM (* 2002 added area from Seven Regions district *) '098' - Portage La Prairie RM '099' - Portage La Prairie City 'A33' - Dakota Tipi FN 'A05' - Dakota Plains FN 'A04' - Long Plain FN Carman (A2C) (* 2002 changed code from A2 to A2C *) '034' - Carman Town '033' - Dufferin RM '053' - Grey RM '112' - Roland RM '195' - St. Claude Village '158' - Thompson RM 1 Manitoba Regional Health Authority (RHA) DISTRICTS MCHP Area -
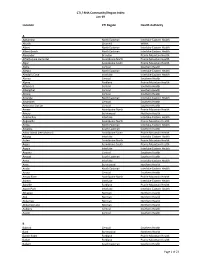
CTI / RHA Community/Region Index Jan-19
CTI / RHA Community/Region Index Jan-19 Location CTI Region Health Authority A Aghaming North Eastman Interlake-Eastern Health Akudik Churchill WRHA Albert North Eastman Interlake-Eastern Health Albert Beach North Eastman Interlake-Eastern Health Alexander Brandon Prairie Mountain Health Alfretta (see Hamiota) Assiniboine North Prairie Mountain Health Algar Assiniboine South Prairie Mountain Health Alpha Central Southern Health Allegra North Eastman Interlake-Eastern Health Almdal's Cove Interlake Interlake-Eastern Health Alonsa Central Southern Health Alpine Parkland Prairie Mountain Health Altamont Central Southern Health Albergthal Central Southern Health Altona Central Southern Health Amanda North Eastman Interlake-Eastern Health Amaranth Central Southern Health Ambroise Station Central Southern Health Ameer Assiniboine North Prairie Mountain Health Amery Burntwood Northern Health Anama Bay Interlake Interlake-Eastern Health Angusville Assiniboine North Prairie Mountain Health Anola North Eastman Interlake-Eastern Health Arbakka South Eastman Southern Health Arbor Island (see Morton) Assiniboine South Prairie Mountain Health Arborg Interlake Interlake-Eastern Health Arden Assiniboine North Prairie Mountain Health Argue Assiniboine South Prairie Mountain Health Argyle Interlake Interlake-Eastern Health Arizona Central Southern Health Amaud South Eastman Southern Health Ames Interlake Interlake-Eastern Health Amot Burntwood Northern Health Anola North Eastman Interlake-Eastern Health Arona Central Southern Health Arrow River Assiniboine -

Section M: Community Support
Section M: Community Support Page 251 of 653 Community Support Health Canada’s Regional Advisor for Children Special Services has developed the Children’s Services Reference Chart for general information on what types of health services are available in the First Nations’ communities. Colour coding was used to indicate where similar services might be accessible from the various community programs. A legend that explains each of the colours /categories can be found in the centre of chart. By using the chart’s colour coding system, resource teachers may be able to contact the communities’ agencies and begin to open new lines of communication in order to create opportunities for cost sharing for special needs services with the schools. However, it needs to be noted that not all First Nations’ communities offer the depth or variety of the services described due to many factors (i.e., budgets). Unfortunately, there are times when special needs services are required but cannot be accessed for reasons beyond the school and community. It is then that resource teachers should contact Manitoba’s Regional Advisor for Children Special Services to ask for direction and assistance in resolving the issue. Manitoba’s Regional Advisor, Children’s Special Services, First Nations and Inuit Health Programs is Mary L. Brown. Phone: 204-‐983-‐1613 Fax: 204-‐983-‐0079 Email: [email protected] On page two is the Children’s Services Reference Chart and on the following page is information from the chart in a clearer and more readable format including -
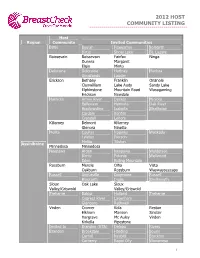
2012 Host Community Listing
2012 HOST COMMUNITY LISTING Host Region Community Invited Communities Birtle Beulah Foxwarren Solsgirth Birtle Shoal Lake St. Lazare Boissevain Boissevain Fairfax Ninga Dunrea Margaret Elgin Minto Deloraine Deloraine Hartney Medora Goodlands Lauder Erickson Bethany Franklin Onanole Clanwilliam Lake Audy Sandy Lake Elphinstone Mountain Road Wasagaming Erickson Newdale Hamiota Arrow River Decker Miniota Belleview Hamiota Oak River Bradwardine Isabella Strathclair Cardale Kenton Crandall Lenore Killarney Belmont Killarney Glenora Ninette Melita Coulter Napinka Waskada Lyleton Pierson Melita Tilston Assiniboine Minnedosa Minnedosa Neepawa Arden Neepawa Waldersee Birnie Polonia Wellwood Eden Riding Mountain Rossburn Menzie Olha Vista Oakburn Rossburn Waywayseecapo Russell Angusville Dropmore Russell Binscarth Inglis Shellmouth Sioux Oak Lake Sioux Valley/Griswold Valley/Griswold Treherne Baldur Holland Treherne Cypress River Lavenham Glenboro Rathwell Virden Cromer Kola Reston Elkhorn Manson Sinclair Hargrave Mc Auley Virden Kirkella Pipestone Invited to Brandon (R7A) Deleau Rivers Brandon Brookdale Harding Souris Carroll Nesbitt Stockton Carberry Rapid City Wawanesa 1 2012 HOST COMMUNITY LISTING Host Region Community Invited Communities Invited to Cartwright Holmfield Mather Assiniboine Crystal City (cont.) Invited to Glenella Kelwood McCreary Churchill Churchill Cross Lake Cross Lake Gillam Gillam Ilford Shamattawa Leaf Rapids Leaf Rapids South Indian Lake Lynn Lake Brochet Lynn Lake Lac Brochet Tadoule Lake Burntwood Nelson House Nelson -

Milk Delivery Services: Rural Manitoba
Milk Delivery Services: Rural Manitoba Milk Delivery Service Delivery Area Brand of Milk Available Arctic Beverages North of 53 Parallel Beatrice Phone: 1-866-503-1270 Eugene Dick Headingly Dairyland Phone: 204-284-5943 Email: [email protected] Gil Marion East & West St. Paul Beatrice Phone: 204-999-2262 Email: [email protected] Grant DeVries Oakbank Beatrice Phone: 204-229-5799 Dugald Email: [email protected] Tyndall Heritage CO-OP Brandon Dairyland Contact: Grocery Department Phone: 204-727-5660 Milk Man Distributors Winnipeg and surrounding area Beatrice Steven Benne Phone : 204-797-7439 [email protected] My Milkman (HDS) Winnipeg & surrounding area Beatrice Phone: 204-777-7000 Email: [email protected] Note: Dairy Farmers of Manitoba is not affiliated with the milk delivery services above. For customer service inquiries please contact the services directly . 1 Milk Delivery Services: Rural Manitoba Milk Delivery Service Delivery Area Brand of Milk Available The North West Berens River Pauingassi Company Brochet Pinawa Churchill Poplar River Contact local in-store Cross Lake Red Sucker Lake Manager Garden Hill Rossville Gods Narrows St. Theresa Point Gods River Shamattawa Lac Brochet South Indian Little Grand Rapids Lake Lynn Lake Split Lake Moose Lake Tadoule Lake Norway House Wasagamack Oxford House Sapotaweyak Pukatawagan Pratt’s Wholesale Ltd Northern MB: Beatrice (North) North of Brandon up to Thompson Darcy Scheller Phone: 204-622-7726 Email: [email protected] Taz Enterprises Ltd South East MB: Beatrice Phone: 204-433-7993 Vita Other: 1-877-811-7880 Ile des Chenes Email: [email protected] St. Pierre Jolys St. Anne Morden Mitchell Landmark Udder One Enterprises South East MB: Lucerne Tom Tomko Dugald Phone: 204-999-6540 Lorette Pager: 204-935-1278 St. -

Sayisi Dene Mapping Initiative, Tadoule Lake Area, Manitoba (Part of NTS 64J9, 10, 15, 16) by L.A
GS-15 Sayisi Dene Mapping Initiative, Tadoule Lake area, Manitoba (part of NTS 64J9, 10, 15, 16) by L.A. Murphy and A.R. Carlson Murphy, L.A. and Carlson, A.R. 2009: Sayisi Dene Mapping Initiative, Tadoule Lake area, Manitoba (part of NTS 64J9, 10, 15, 16); in Report of Activities 2009, Manitoba Innovation, Energy and Mines, Manitoba Geological Survey, p. 154–159. Summary metasedimentary units and rafts In the summer of 2009, the Manitoba Geological Sur- of possible Archean provenance. vey (MGS) conducted the first half of a four-week geo- Petrology, geochemistry and Sm- logical mapping course with the Sayisi Dene First Nation Nd isotope analyses of samples at Tadoule Lake, Manitoba. The goals of the program are collected in the Tadoule Lake area will provide constraints to help increase awareness of the region’s geology and necessary to determine the nature, ages and contact rela- mineral resources, provide the basic skills needed to work tionships of exposed rock types. This work may help in a mineral-exploration camp and foster information- interpret geological domain boundaries in the Tadoule sharing between First Nation communities and the MGS. Lake area in conjunction with other projects that are all Four members of the community were hired as student- part of the ongoing Manitoba Far North Geomapping Ini- trainees and three completed the first half of the program. tiative (Anderson, et al., GS-13, this volume). One of the trainees was hired by the MGS as a field assis- tant to geologists working in the Seal River–Great Island Introduction area between Tadoule Lake and Churchill. -

RELOCATION and LOSS of HOMELAND the Story of the Sayisi Dene of Northern Manitoba
RELOCATION AND LOSS OF HOMELAND THE STORY OF THE SAYIS'I DENE: OF NORTEERN MANITOBA BY VIRGINIA PHYLLIS PETCH A Thesis presented to the University of Manitoba in partial fulfillment of the requirements of a Doctor of Philosophy in Anthropology The University of Manitoba Winnipeg, Manitoba June, 1998 O Copyright Viginia P. Petch, 1998. Ail rights reserved National Library Bibliothèque nationale u*m of Carda du Canada Acquisitions and Acquisitions et Bibliographie SeMces seMces bibliographiques 395 WMingtOCI Street 395, nie Wdtington OtîawaON K1AûN4 OttawaON K1A ON4 canada canada The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/film, de reproduction sur papier ou sur format électronique. The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantid extracts from it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation. FACULTY OF GWUATE STLDIES ***** COPYRIGHT PER\LISSIO?i PAGE A Thesis/Practicum submitted to the Facuky of Graduate Shidies of The University of SIanitoba in partial fulfillmeat of the requirements of the degree of Permission has been granted to the Library of The University of Manitoba to lend or seii copies of this thesidpracticum, to the National Library of Canada <O microfilm thQ thesis and to lend or sel1 copies of the fdm, and to Dissertations Absmcts International to publish an abstract of this thesis/practicum. -

Optimizing an Off-Grid Electrical System in Brochet, Manitoba, Canada Renewable and Sustainable Energy Reviews
Renewable and Sustainable Energy Reviews 53 (2016) 709–719 Contents lists available at ScienceDirect Renewable and Sustainable Energy Reviews journal homepage: www.elsevier.com/locate/rser Optimizing an off-grid electrical system in Brochet, Manitoba, Canada Prasid Ram Bhattarai n, Shirley Thompson Natural Resources Institute, University of Manitoba, Canada R3T 2M6 article info abstract Article history: Brochet is a remote, off-grid community located in Northern Manitoba, Canada. The existing diesel Received 28 October 2014 generating system is characterized by high economic and environmental cost. As the existing diesel Received in revised form generators are nearing their operational lifespan, this study uses the HOMER model to determine an 4 July 2015 optimum electricity system design at Brochet that has high electrical reliability, least cost, and low Accepted 1 September 2015 emissions. Two potential power generation options based on reduced sized diesel generator, and a Available online 30 September 2015 wind–diesel hybrid system were evaluated and compared against the existing diesel-based electricity Keywords: system at Brochet. The wind–diesel hybrid system performed best in all three (i.e. electrical, economics, HOMER model and environmental) evaluation criteria. While maintaining high reliability, this hybrid system design Off-grid resulted in 30% reduction in cost of electricity produced, and 18% reduction of carbon dioxide emissions Optimization when compared to the existing electricity system at Brochet. Thus, this study concludes that the wind– Wind–diesel hybrid diesel hybrid system is the optimum electricity system design for Brochet and proposes this system to replace the existing system. & 2015 Elsevier Ltd. All rights reserved. Contents 1. -

Future Regional Resource Development
FUTURE REGIONAL RESOURCE DEVELOPMENT In this section: 1. AGRICULTURE ........................................................................................ 1 2. FORESTRY .............................................................................................. 5 3. MINING and METAL PROCESSING or HANDLING .................................... 7 4. ENERGY ............................................................................................... 14 5. MANUFACTURING ............................................................................... 20 AGRICULTURE 1. AGRICULTURE The State of Agriculture in Manitoba, a report provided by the Manitoba government 1, provides a comprehensive overview of the agricultural industry, both currently and historically. Key highlights from this report include: • The agricultural industry in Manitoba is well established, although a relatively small part of the economy. On average, agricultural production contributes about 4.5 percent to the total provincial gross domestic product (GDP), food processing contributes an additional 2 to 4 percent. • The amount of farmland in Manitoba in 2011 was slightly lower than in 2006 (18 million acres compared to 19.1 million acres), but in general, the amount of farmland has remained relatively stable over the past 30 years (Figure 1.1) 1. • The top five activities producing the most in terms of farm cash receipts are canola, hogs, wheat, cattle and dairy (Figure 1.2) 1. • Canola production has grown steadily and accounts for the largest amount of seeded area. -

Regional Environment Offices – Contact and Coverage Information
Regional Environment Offices – Contact and Coverage Information Winnipeg Office – (204) 945-0675 Portage la Prairie Office – (204) 239-3608 1007 Century Street, R3H 0W4 309 - 25 Tupper Street North, R1N 3K1 City of Winnipeg RM of Dufferin RM of Cartier RM of Grey RM of Headingley RM of Lorne RM of MacDonald RM of Louise RM of Rosser RM of Montcalm RM of St. François Xavier RM of Morris RM of East St. Paul RM of Norfolk-Treherne RM of Ritchot RM of North Norfolk RM of Springfield RM of Pembina RM of Portage la Prairie RM of Rhineland Selkirk Office – (204) 785-5030 RM of Roland 446 Main Street, Lower Level, R1A 1V7 RM of Stanley RM of Thompson RM of Coldwell RM of Victoria RM of Grahamdale RM of WestLake-Gladstone RM of Rockwood Stephenfield Provincial Park RM of St. Andrews Pembina Valley Provincial Park RM of St. Clements RM of St. Laurent RM of Victoria Beach Lac du Bonnet Office – (204) 345-1490 RM of West Interlake Provincial Hwy 502, Box 4000, R0E 1A0 RM of Woodlands Village of Dunnottar RM of Alexander City of Selkirk RM of Brokenhead Town of Winnipeg Beach RM of Lac du Bonnet Birds Hill Provincial Park LGD of Pinawa Grand Beach Provincial Park RM of Reynolds Matheson Island RM of Whitemouth Pine Dock Whiteshell Provincial Park (North) Nopoming Provincial Park Atikaki Provincial Park Gimli Office – (204) 641-4091 Manigotagan 75 – 7th Avenue, Box 6000, ROC 1B0 Town of Powerview-Pine Falls Seymourville RM of Armstrong Town of Beausejour RM of Bifrost-Riverton Bisset RM of -

Central Eastman Interlake Norman Parkland Westman Winnipeg Altamont Anola Arborg Big Eddy Alsona Baldur District # 1 Altona Arna
Up-dated May 26, 2008 Central Eastman Interlake Norman Parkland Westman Winnipeg Altamont Anola Arborg Big Eddy Alsona Baldur District # 1 Altona Arnaud Argyle Brochet Amaranth Beulah Clifton C.C. Arden Beausejour Arnes Churchill Angusville Beulah Crescentwood C.C. Austin Berens River Ashern Cormorant Barrows Birtle Earl Grey C.C. Brunkild Bisset Balmoral Cranberry Portage Bellsite Boissevain Fort Garry C.C. Bruxelles Cook's Creek Beconia Cross Lake Benito Brandon Isaac Brock C.C. Carman Dominion City Birds Hill Denare Beach, SK Binscarth Carberry Lord Roberts C.C. Cartwright Dufrost Camp Morton Easterville Birch River Deloraine Orioles C.C. Clearwater Dugald Chatfield Flin Flon Bowsman Elkhorn River Heights C.C. Crystal City Elma Clandeboye Garden Hill Camperville Erickson River Osborne C.C. Cypress River Falcon Lake East Selkirk Gillam Cowan Foxwarren Riverview C.C. Domain Garson East St. Paul God's Lake Narrows Crane River Glenboro Robert A. Steen Memorial C.C. Elie Grande Pointe Eriksdale God's River Dauphin Glenora Sir John Franklin C.C. Elmcreek Great Falls Fisherbranch Grand Rapids Ethelbert Hamiota Victoria-Linden Woods C.C. Emerson Hadashville Fraserwood Granville Lake Fork River Hartney Westridge C.C. Fannystelle Hazelridge Gimili Herb Lake Landing Gilbert Plains Kenton Wildwood C.C. Gladstone Ile Des Chenes Grand Marias Ilford Grandview Killarney Glenella Grosse Isle Lac Brochet Inglis Lenore District # 2 Gretna La Broquerie Gympsumville Leaf Rapids Laurier McAuley Assiniboia West C.C. Halbstadt Lac du Bonnet Hodgson Lynn Lake Mafeking Melita Bord-Aire C.C. Haywood Landmark Inwood Moose Lake McCreary Miniota Bourkevale C.C. Headingly Lorette Kirkness Nelson House Minitonas Minnedosa Brooklands C.C.