DNA Methylation Ageing Clocks: Challenges & Recommendations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cognitive Epidemiology: Its Rise, Its Current Issues, and Its Challenges
Edinburgh Research Explorer Cognitive epidemiology: Its rise, its current issues, and its challenges Citation for published version: Deary, IJ 2010, 'Cognitive epidemiology: Its rise, its current issues, and its challenges', Personality and Individual Differences, vol. 49, no. 4, pp. 337-343. https://doi.org/10.1016/j.paid.2009.11.012 Digital Object Identifier (DOI): 10.1016/j.paid.2009.11.012 Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Personality and Individual Differences Publisher Rights Statement: This is an Author's Accepted Manuscript of the following article: Deary, I. J. (2010) "Cognitive epidemiology: Its rise, its current issues, and its challenges", in Personality and Individual Differences. 49, 4, p. 337-343. © Elsevier. The final publication is available at http://dx.doi.org/10.1016/j.paid.2009.11.012 General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 1 Cognitive epidemiology: its rise, its current issues, and its challenges Ian J. Deary Centre for Cognitive Ageing and Cognitive Epidemiology, Department of Psychology, University of Edinburgh, Scotland, United Kingdom Correspondence: Ian J. -

When Psychometrics Meets Epidemiology and the Studies V1.Pdf
When psychometrics meets epidemiology, and the studies which result! Tom Booth [email protected] Lecturer in Quantitative Research Methods, Department of Psychology, University of Edinburgh Who am I? • MSc and PhD in Organisational Psychology – ESRC AQM Scholarship • Manchester Business School, University of Manchester • Topic: Personality psychometrics • Post-doctoral Researcher • Centre for Cognitive Ageing and Cognitive Epidemiology, Department of Psychology, University of Edinburgh. • Primary Research Topic: Cognitive ageing, brain imaging. • Lecturer Quantitative Research Methods • Department of Psychology, University of Edinburgh • Primary Research Topics: Individual differences and health; cognitive ability and brain imaging; Psychometric methods and assessment. Journey of a talk…. • Psychometrics: • Performance of likert-type response scales for personality data. • Murray, Booth & Molenaar (2015) • Epidemiology: • Allostatic load • Measurement: Booth, Starr & Deary (2013); (Unpublished) • Applications: Early life adversity (Unpublished) • Further applications Journey of a talk…. • Methodological interlude: • The issue of optimal time scales. • Individual differences and health: • Personality and Physical Health (review: Murray & Booth, 2015) • Personality, health behaviours and brain integrity (Booth, Mottus et al., 2014) • Looking forward Psychometrics My spiritual home… Middle response options Strongly Agree Agree Neither Agree nor Disagree Strong Disagree Disagree 1 2 3 4 5 Strongly Agree Agree Unsure Disagree Strong Disagree -

Independent Evidence for an Association Between General Cognitive Ability and a Genetic Locus for Educational Attainment J
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Hofstra Northwell Academic Works (Hofstra Northwell School of Medicine) Donald and Barbara Zucker School of Medicine Journal Articles Academic Works 2015 Independent evidence for an association between general cognitive ability and a genetic locus for educational attainment J. W. Trampush Hofstra Northwell School of Medicine T. Lencz Hofstra Northwell School of Medicine S. Guha Northwell Health S. Mukherjee Northwell Health P. DeRosse Northwell Health See next page for additional authors Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Psychiatry Commons Recommended Citation Trampush JW, Lencz T, Guha S, Mukherjee S, DeRosse P, John M, Andreassen O, Deary I, Glahn D, Malhotra AK, . Independent evidence for an association between general cognitive ability and a genetic locus for educational attainment. 2015 Jan 01; 168(5):Article 927 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/927. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. Authors J. W. Trampush, T. Lencz, S. Guha, S. Mukherjee, P. DeRosse, M. John, O. A. Andreassen, I. J. Deary, D. C. Glahn, A. K. Malhotra, and +41 additional authors This article is available at Donald and Barbara Zucker School of Medicine Academic Works: https://academicworks.medicine.hofstra.edu/articles/927 HHS Public Access Author manuscript Author Manuscript Author ManuscriptAm J Med Author Manuscript Genet B Neuropsychiatr Author Manuscript Genet. -

Cognitive Ageing and Cognitive Epidemiology
Centre for Cognitive Ageing and Cognitive Epidemiology Cognitive decline is the single most feared aspect of growing old. It is also the most costly, in terms of the financial, the personal and societal burden. Many organisations—governmental, academic, research-funding, professional and charitable—have announced that is has to be addressed, that too little progress has been made in the field, and that more integrated research is needed to understand the mechanisms of cognitive ageing . The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology will answer this call directly by working on identifying the risk factors for, and mechanisms of, individual differences in age-related cognitive decline—cognitive ageing—and its relationship to morbidity and mortality in later life—cognitive epidemiology. The Centre’s objectives The Centre will initially run from 2008 to 2013. It will enhance the University of Edinburgh’s research on lifelong health and wellbeing, especially with regard to cognition. It will have a doctoral training programme and clinical research fellowship. The Centre’s objectives are: • To explore lifecourse influences on cognitive ageing and pathways whereby cognitive ability in early life affects later health—cognitive epidemiology. As a contribution to this it will maintain, develop and exploit the unique long-term human cohort studies assembled in Scotland as new national resources. • To advance knowledge by research into biological, neurological, genetic, social, economic, and psychological aspects of cognitive ageing in humans and lifecourse mammalian model systems. • To develop and evaluate methods in psychological, genetic, other biological, and brain imaging science to assess, monitor, and prevent or ameliorate decline in mental functions with a view to providing a rational basis for translating this into potential interventions. -

Epigenetic Age Acceleration and Cognitive Function in African
Edinburgh Research Explorer Epigenetic age acceleration and cognitive function in African- American adults in midlife Citation for published version: Bressler, J, Marioni, RE, Walker, RM, Xia, R, Gottesman, RF, Windham, BG, Grove, ML, Guan, W, Pankow, JS, Evans, KL, Mcintosh, AM, Deary, IJ, Mosley, TH, Boerwinkle, E & Fornage, M 2020, 'Epigenetic age acceleration and cognitive function in African-American adults in midlife: The Atherosclerosis Risk in Communities Study', Journal of Gerontology: Medical Sciences, vol. 75, no. 3, glz245, pp. 473–480. https://doi.org/10.1093/gerona/glz245 Digital Object Identifier (DOI): 10.1093/gerona/glz245 Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Journal of Gerontology: Medical Sciences Publisher Rights Statement: This is a pre-copyedited, author-produced version of an article accepted for publication in Journals of Gerontology, Series A following peer review. The version of record: Jan Bressler, Riccardo E Marioni, Rosie M Walker, Rui Xia, Rebecca F Gottesman, B Gwen Windham, Megan L Grove, Weihua Guan, James S Pankow, Kathryn L Evans, Andrew M McIntosh, Ian J Deary, Thomas H Mosley, Eric Boerwinkle, Myriam Fornage, Epigenetic Age Acceleration and Cognitive Function in African-American Adults in Midlife: The Atherosclerosis Risk in Communities Study, The Journals of Gerontology: Series A, , glz245 is available online at: https://doi.org/10.1093/gerona/glz245 General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. -

2019 NACA BSR Review Committee Report
National Institute on Aging (NIA) National Advisory Council on Aging (NACA) Review of the Division of Behavioral and Social Research (BSR) 2019 BSR Review Committee Report Rev. December 16, 2019 Review Committee Members (*current NIA Council Member, **former NIA Council Member) *Eileen Crimmins, PhD, University of Southern California (Co-Chair) *Terrie Moffitt, PhD, Duke University and King’s College, London (Co-Chair) *David Bennett, MD, Rush University **Lisa Berkman, PhD, Harvard University Anne Case, PhD, Princeton University Michael Chernew, PhD, Harvard University Elissa Epel, PhD, University of California, San Francisco Margaret Gatz, PhD, University of Southern California Maria Glymour, University of California, San Francisco *J Taylor Harden, PhD, RN, FGSA, FAAN, National Hartford Center of Gerontological Nursing Excellence Arthur Kramer, PhD, Northeastern University *Stephen Kritchevsky, PhD, Wake Forest School of Medicine Robert Levenson, PhD, University of California, Berkeley Jennifer Manly, PhD, Columbia University Vincent Mor, PhD, Brown University Elizabeth Phelps, PhD, Harvard University Kevin Volpp, MD, PhD, University of Pennsylvania Rose Maria Li, MBA, PhD, Executive Secretary NIA BSR Review, January 2020 Rev. 12/16/19 Table of Contents I. Executive Summary ..................................................................................................... 3 A. The Current BSR Research Portfolio .................................................................... 3 B. Recommendations for Future Focus .................................................................. -

Childhood Coordination and Survival up to Six Decades Later: Extended Follow-Up of Participants in the National Child Development Study
medRxiv preprint doi: https://doi.org/10.1101/19004713; this version posted August 13, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Original Investigation Childhood coordination and survival up to six decades later: extended follow-up of participants in the National Child Development Study G. David Batty,a,b PhD, DSc (E. [email protected]) Ian J. Deary,c PhD (E. [email protected]) Mark Hamer,d PhD (E. [email protected]) Stuart J. Ritchie,e PhD (E. [email protected]) David Bann,f PhD (E. [email protected]) aDepartment of Epidemiology & Public Health, University College London, UK bSchool of Biological & Population Health Sciences, Oregon State University, USA cCentre for Cognitive Ageing & Cognitive Epidemiology, University of Edinburgh, UK dSchool of Sport, Exercise & Health Sciences, Loughborough University, UK eSocial, Genetic & Developmental Psychiatry Centre, King’s College London, UK fCentre for Longitudinal Studies, University College London Institute of Education, UK Correspondence: David Batty, Department of Epidemiology & Public Health, University College London, 1-19 Torrington Place, London, UK, WC1E 6BT Manuscript statistics: 2969 words (main text), 1 table, 4 figures, 1 supplemental file Funding: GDB is supported by the UK Medical Research Council (MR/P023444/1) and the US National Institute on Aging (1R56AG052519-01; 1R01AG052519-01A1), DB by the Economic and Social Research Council (grant numbers ES/M001660/1) and The Academy of Medical Sciences/Wellcome Trust (HOP001/1025). -

Encyclopedia of Geropsychology This Is a FM Blank Page Nancy A
Encyclopedia of Geropsychology ThiS is a FM Blank Page Nancy A. Pachana Editor Encyclopedia of Geropsychology With 148 Figures and 100 Tables Editor Nancy A. Pachana The University of Queensland Brisbane, QLD, Australia ISBN 978-981-287-081-0 ISBN 978-981-287-082-7 (eBook) ISBN 978-981-287-083-4 (print and electronic bundle) DOI 10.1007/978-981-287-082-7 Library of Congress Control Number: 2016953014 # Springer Science+Business Media Singapore 2017 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the editors give a warranty, express or implied, with respect to the material contained herein or for any errors or omissions that may have been made. Printed on acid-free paper This Springer imprint is published by Springer Nature The registered company is Springer Nature Singapore Pte Ltd. -

Identification of Genetic Loci Jointly Influencing Schizophrenia Risk And
Research JAMA Psychiatry | Original Investigation Identification of Genetic Loci Jointly Influencing Schizophrenia Risk and the Cognitive Traits of Verbal-Numerical Reasoning, Reaction Time, and General Cognitive Function Olav B. Smeland, MD, PhD; Oleksandr Frei, PhD; Karolina Kauppi, PhD; W. David Hill, PhD; Wen Li, PhD; Yunpeng Wang, PhD; Florian Krull, PhD; Francesco Bettella, PhD; Jon A. Eriksen, PhD; Aree Witoelar, PhD; Gail Davies, PhD; Chun C. Fan, MD; Wesley K. Thompson, PhD; Max Lam, PhD; Todd Lencz, PhD; Chi-Hua Chen, PhD; Torill Ueland, PhD; Erik G. Jönsson, MD, PhD; Srdjan Djurovic, PhD; Ian J. Deary, PhD; Anders M. Dale, PhD; Ole A. Andreassen, MD, PhD; for the NeuroCHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) Cognitive Working Group Supplemental content IMPORTANCE Schizophrenia is associated with widespread cognitive impairments. Although cognitive deficits are one of the factors most strongly associated with functional outcome in schizophrenia, current treatment strategies largely fail to ameliorate these impairments. To develop more efficient treatment strategies in patients with schizophrenia, a better understanding of the pathogenesis of these cognitive deficits is needed. Accumulating evidence indicates that genetic risk of schizophrenia may contribute to cognitive dysfunction. OBJECTIVE To identify genomic regions jointly influencing schizophrenia and the cognitive domains of reaction time and verbal-numerical reasoning, as well as general cognitive function, a phenotype that captures the shared variation in performance across cognitive domains. DESIGN, SETTING, AND PARTICIPANTS Combining data from genome-wide association studies from multiple phenotypes using conditional false discovery rate analysis provides increased power to discover genetic variants and could elucidate shared molecular genetic mechanisms. -
Dong Et Al. Reply Replying to N
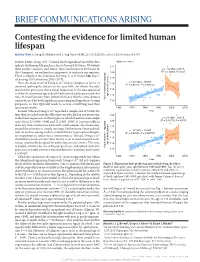
BRIEF COMMUNICATIONS ARISING Contesting the evidence for limited human lifespan ARISING FROM X. Dong, B. Milholland & J. Vijg Nature 538, 257–259 (2016); doi:10.1038/nature19793 In their Letter, Dong et al.1 claimed that longitudinal mortality data Figure 2a of ref. 1 indicate that human lifespan has a limit of around 115 years. We believe 122 these authors’ analyses, and, hence, their conclusions to be flawed. In y = –0.2762x + 667.5 2 this Comment, we outline four arguments to motivate our opinion. 120 R = 0.12001, P = 0.27 There is a Reply to this Comment by Dong, X. et al. Nature 546, http:// 118 dx.doi.org/10.1038/nature22785 (2017). 1 y = 0.1531x – 191.07 116 First, the main result of Dong et al. (shown in figure 2a of ref. 1) R2 = 0.45855, P = 0.000747 involved splitting the dataset at the year 1995, for which the only justification given was that a visual inspection of the data appeared 114 to show the maximum age at death had reached a plateau around that 112 time. It is well-known from statistical theory that the same dataset age at death (years) cannot be used for both hypothesis-generating and hypothesis-testing Yearly maximum reported 110 purposes, as this typically leads to severe overfitting and thus inaccurate results. 1960 1970 1980 1990 2000 2010 Second, whereas Dong et al.1 reported a sample size of 534 in the Year text, they included only the oldest person who died in any given year 122 y = 0.1399x – 164.79 in the linear regressions in their figure 2a, which therefore used sample 2 sizes of just 21 (1968–1994) and 12 (1995–2006). -

A Comment on “Fractionating Intelligence” and the Peer Review Process
Intelligence 46 (2014) 323–332 Contents lists available at ScienceDirect Intelligence A comment on “Fractionating Intelligence” and the peer review process Richard J. Haier a,⁎, Sherif Karama b,c, Roberto Colom d, Rex Jung e, Wendy Johnson f a School of Medicine (Emeritus), University of California, Irvine, CA, USA b McConnell Brain Imaging Center, Montreal Neurological Institute, McGill University, Montreal, Canada c Douglas Mental Health University Institute, Department of Psychiatry, McGill University, Montreal, Canada d Universidad Autónoma de Madrid-Fundación CIEN/Fundación Reina Sofía, Spain e Department of Neurosurgery, University of New Mexico, Albuquerque, NM, USA f MRC Centre for Cognitive Ageing and Cognitive Epidemiology, Department of Psychology, University of Edinburgh, UK article info abstract Article history: Hampshire and colleagues used factor analyses and simulations to conclude that the g-factor is Received 6 February 2014 not a valid construct for general intelligence because it could be accounted for by at least two Received in revised form 18 February 2014 independent components defined by distinct brain networks. In our view, their results depend Accepted 20 February 2014 on a number of assumptions and subjective decisions that, at best, allow for different Available online 6 April 2014 interpretations. We also had a unique role in the review process of their paper prior to its publication when we were invited to write a Preview. We detail that role here and describe Keywords: how non-transparent editorial decision-making rejected our Preview and allowed publication g-Factor despite our major concerns. The main purpose of this report is to invite Hampshire and Intelligence colleagues to respond to our specific scientific concerns that aim to clarify their work and Factor analysis contribute a constructive discussion about the meaning of their findings. -

Influencing the Trajectories of Ageing
Influencing the trajectories of ageing Summary of a FORUM symposium held on 16 September 2016 The Academy of Medical Sciences The Academy of Medical Sciences is the independent body in the UK representing the diversity of medical science. Our mission is to promote medical science and its translation into benefits for society. The Academy’s elected Fellows are the United Kingdom’s leading medical scientists from hospitals, academia, industry and the public service. We work with them to promote excellence, influence policy to improve health and wealth, nurture the next generation of medical researchers, link academia, industry and the NHS, seize international opportunities and encourage dialogue about the medical sciences. The Academy of Medical Sciences’ FORUM The Academy’s FORUM was established in 2003 to recognise the role of industry in medical research, and to catalyse connections across industry, academia and the NHS. Since then, a range of FORUM activities and events have brought together researchers, research funders and research users from across academia, industry, government, and the charity, healthcare and regulatory sectors. The FORUM is a major component of the Academy's work to deliver the strategic objective of 'linking academia, industry and the NHS' and its success relies on supporter organisations who make an annual donation. We are grateful for the support provided by the members and are keen to encourage more organisations to take part. If you would like further information on the FORUM or becoming a member, please contact [email protected]. Opinions expressed in this report do not necessarily represent the views of all participants at the event, the Academy of Medical Sciences, or its Fellows.