Thermodynamics of Concentrated Solid Solution Alloys ⇑ M.C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DFT Quantization of the Gibbs Free Energy of a Quantum Body
DFT quantization of the Gibbs Free Energy of a quantum body S. Selenu (Dated: December 2, 2020) In this article it is introduced a theoretical model made in order to perform calculations of the quantum heat of a body that could be acquired or delivered during a thermal transformation of its quantum states. Here the model is mainly targeted to the electronic structure of matter[1] at the nano and micro scale where DFT models have been frequently developed of the total energy of quantum systems. De fining an Entropy functional S[ρ] makes us able optimizing a free energy G[ρ] of the quantum system at finite temperatures. Due to the generality of the model, the latter can be also applied to several first principles computational codes where ab initio modelling of quantum matter is asked. INTRODUCTION thermal transformations at a fi nite temperature T . This work could then be consid- This article will be focused on a study based on the ered as the begining part of a more general theory can derivation of the quantum electrical heat absorbed or ei- include an ab initio modelling of the first law and second ther released by a physical system made of atoms and law of thermodynamics[13], where heat and free energy electrons either at the nano or at the microscopic scale, variations of a quantum body are taken into account. In extending to it the concept of thermal heat within a DFT fact it is actually considered a quantum system of elec- model. Also, the latter being fundamental in physics, can trons at a given temperature T in thermal equilibrium be employed for a full understanding of the thermal phase with its surrounding. -
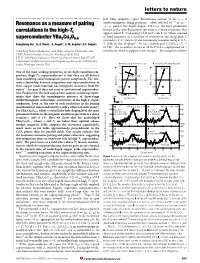
Resonance As a Measure of Pairing Correlations in the High-Tc
letters to nature ................................................................. lost while magnetic (spin) ¯uctuations centred at the x =0 6±11 Resonance as a measure of pairing antiferromagnetic Bragg positionsÐoften referred to as Q0 = (p, p)±persist. For highly doped (123)O6+x, the most prominent feature in the spin ¯uctuation spectrum is a sharp resonance that correlations in the high-Tc appears below Tc at an energy of 41 meV (refs 6±8). When scanned superconductor YBa Cu O at ®xed frequency as a function of wavevector, the sharp peak is 2 3 6.6 centred at (p, p) (refs 6±8) and its intensity is unaffected by a 11.5- 19 Pengcheng Dai*, H. A. Mook*, G. Aeppli², S. M. Hayden³ & F. DogÏan§ T ®eld in the ab-plane . In our underdoped (123)O6.6 (Tc = 62.7 K)9, the resonance occurs at 34 meV and is superposed on a * Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831-6393, USA continuum which is gapped at low energies11. For frequencies below ² NEC Research Institute, Princeton, New Jersey 08540, USA ³ H. H. Wills Physics Laboratory, University of Bristol, Bristol BS8 1TL, UK § Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA B rotated 20.6° from the c-axis B along the [3H,-H,0] direction 2.0 2.0 .............................................................................................................................................. a d [H,3H,0] One of the most striking properties of the high-transition-tem- 1.5 1.5 perature (high-Tc) superconductors is that they are all derived [H, (5-H)/3, 0] from insulating antiferromagnetic parent compounds. The inti- 1.0 B ∼ i c-axis 1.0 B i ab-plane mate relationship between magnetism and superconductivity in these copper oxide materials has intrigued researchers from the [0,K,0] (r.l.u.) [0,K,0] (r.l.u.) 0.5 0.5 outset1±4, because it does not exist in conventional superconduc- 20.6° tors. -

A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Article A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action Ivan Kennedy 1,2,*, Harold Geering 2, Michael Rose 3 and Angus Crossan 2 1 Sydney Institute of Agriculture, University of Sydney, NSW 2006, Australia 2 QuickTest Technologies, PO Box 6285 North Ryde, NSW 2113, Australia; [email protected] (H.G.); [email protected] (A.C.) 3 NSW Department of Primary Industries, Wollongbar NSW 2447, Australia; [email protected] * Correspondence: [email protected]; Tel.: + 61-4-0794-9622 Received: 23 March 2019; Accepted: 26 April 2019; Published: 1 May 2019 Abstract: A convenient practical model for accurately estimating the total entropy (ΣSi) of atmospheric gases based on physical action is proposed. This realistic approach is fully consistent with statistical mechanics, but reinterprets its partition functions as measures of translational, rotational, and vibrational action or quantum states, to estimate the entropy. With all kinds of molecular action expressed as logarithmic functions, the total heat required for warming a chemical system from 0 K (ΣSiT) to a given temperature and pressure can be computed, yielding results identical with published experimental third law values of entropy. All thermodynamic properties of gases including entropy, enthalpy, Gibbs energy, and Helmholtz energy are directly estimated using simple algorithms based on simple molecular and physical properties, without resource to tables of standard values; both free energies are measures of quantum field states and of minimal statistical degeneracy, decreasing with temperature and declining density. We propose that this more realistic approach has heuristic value for thermodynamic computation of atmospheric profiles, based on steady state heat flows equilibrating with gravity. -

Proximity to a Critical Point Driven by Electronic Entropy in Uru2si2
www.nature.com/npjquantmats ARTICLE OPEN Proximity to a critical point driven by electronic entropy in URu2Si2 ✉ Neil Harrison 1 , Satya K. Kushwaha1,2, Mun K. Chan 1 and Marcelo Jaime 1 The strongly correlated actinide metal URu2Si2 exhibits a mean field-like second order phase transition at To ≈ 17 K, yet lacks definitive signatures of a broken symmetry. Meanwhile, various experiments have also shown the electronic energy gap to closely resemble that resulting from hybridization between conduction electron and 5f-electron states. We argue here, using thermodynamic measurements, that the above seemingly incompatible observations can be jointly understood by way of proximity to an entropy-driven critical point, in which the latent heat of a valence-type electronic instability is quenched by thermal excitations across a gap, driving the transition second order. Salient features of such a transition include a robust gap spanning highly degenerate features in the electronic density of states, that is weakly (if at all) suppressed by temperature on approaching To, and an elliptical phase boundary in magnetic field and temperature that is Pauli paramagnetically limited at its critical magnetic field. npj Quantum Materials (2021) 6:24 ; https://doi.org/10.1038/s41535-021-00317-6 1234567890():,; INTRODUCTION no absolute requirement for the magnitude of a hybridization gap URu2Si2 remains of immense interest owing to the possibility to vanish at a phase transition, it need not be thermally of it exhibiting a form of order distinct from that observed -

Electronic Entropy Contribution to the Metal Insulator Transition in VO$ 2
Electronic entropy contribution to the metal insulator transition in VO2 The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Paras, J. and A. Allanore. "Electronic entropy contribution to the metal insulator transition in VO₂" Physical Review B 102, 16 (October 2020): 165138. © 2020 American Physical Society As Published http://dx.doi.org/10.1103/physrevb.102.165138 Publisher American Physical Society (APS) Version Final published version Citable link https://hdl.handle.net/1721.1/131098 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. PHYSICAL REVIEW B 102, 165138 (2020) Electronic entropy contribution to the metal insulator transition in VO2 J. Paras and A. Allanore Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA (Received 11 June 2020; revised 31 August 2020; accepted 1 September 2020; published 21 October 2020) VO2 experiences a metal-insulator transition at 340 K. Discontinuities in electronic transport properties, such as the Seebeck coefficient and the electronic conductivity, suggest that there is a significant change in the electronic structure upon metallization. However, the thermodynamic nature of this transformation remains difficult to describe using conventional computational and experimental methods. This has led to disagreement over the relative importance of the change in electronic entropy with respect to the overall transition entropy. A method is presented that links measurable electronic transport properties to the change in electronic state entropy of conduction electrons. The change in electronic entropy is calculated to be 9.2 ± 0.7J/mol K which accounts for 62%–67% of the total transition entropy. -

Linear Dielectric Thermodynamics
Linear Dielectric Thermodynamics: A New Universal Law for Optical, Dielectric Constants by S. J. Burns Materials Science Program Department of Mechanical Engineering University of Rochester Rochester, NY 14627 USA April 17, 2020 Abstract Linear dielectric thermodynamics are formally developed to explore the isothermal and adiabatic temperature - pressure dependence of dielectric constants. The refractive index of optical materials is widely measured in the literature: it is both temperature and pressure dependent. The argument to establish the dielectric constant’s isentropic temperature dependence is a thermodynamic one and is thus applicable to all physical models that describe electron clouds and electronic resonances within materials. The isentropic slope of the displacement field versus the electric field at all temperatures is described by an adiabatic dielectric constant in an energy- per-unit mass system. This slope is shown through the electronic part of the entropy to be unstable at high temperatures due to the change in the curvature of the temperature dependence of the dielectric constant. The electronic entropy contribution for optical, thermo-electro materials has negative heat capacities which are unacceptable. The dielectric constant’s temperature and pressure dependence is predicted to be only dependent on the specific volume so isentropes are always positive. A new universal form for the dielectric constant follows from this hypothesis: the dielectric constant is proportional to the square root of the specific volume. -

Electronic and Transport Properties of Weyl Semimetals
Electronic and Transport Properties of Weyl Semimetals Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Timothy M. McCormick, B.S. Graduate Program in Physics The Ohio State University 2018 Dissertation Committee: Professor Nandini Trivedi, Advisor Professor Yuri Kovchegov Professor Mohit Randeria Professor Rolando Valdes Aguilar c Copyright by Timothy M. McCormick 2018 Abstract Topological Weyl semimetals have attracted substantial recent interest in con- densed matter physics. In this thesis, we theoretically explore electronic and transport properties of these novel materials. We also present results of experimental collabora- tions that support our theoretical calculations. Topological Weyl semimetals (TWS) can be classified as type-I TWS, in which the density of states vanishes at the Weyl nodes, and type-II TWS, in which an electron pocket and a hole pocket meet at a singular point of momentum space, allowing for distinct topological properties. We consider various minimal lattice models for type-II TWS. We present the discovery of a type II topological Weyl semimetal (TWS) state in pure MoTe2, where two sets of WPs (W2±, W3±) exist at the touching points of electron and hole pockets and are located at different binding energies above EF . Using ARPES, modeling, DFT and calculations of Berry curvature, we identify the Weyl points and demonstrate that they are connected by different sets of Fermi arcs for each of the two surface terminations. Weyl semimetals possess low energy excitations which act as monopoles of Berry curvature in momentum space. These emergent monopoles are at the heart of the extensive novel transport properties that Weyl semimetals exhibit. -

First Principles Calculation of the Entropy of Liquid Aluminum
entropy Article First Principles Calculation of the Entropy of Liquid Aluminum Michael Widom 1,* and Michael Gao 2,3 1 Department of Physics, Carnegie Mellon University, Pittsburgh, PA 15217, USA 2 National Energy Technology Laboratory, Albany, OR 97321, USA; [email protected] 3 Leidos Research Support Team, P.O. Box 10940, Pittsburgh, PA 15236, USA * Correspondence: [email protected]; Tel.: +1-412-268-7645 Received: 5 December 2018; Accepted: 23 January 2019; Published: 31 January 2019 Abstract: The information required to specify a liquid structure equals, in suitable units, its thermodynamic entropy. Hence, an expansion of the entropy in terms of multi-particle correlation functions can be interpreted as a hierarchy of information measures. Utilizing first principles molecular dynamics simulations, we simulate the structure of liquid aluminum to obtain its density, pair and triplet correlation functions, allowing us to approximate the experimentally measured entropy and relate the excess entropy to the information content of the correlation functions. We discuss the accuracy and convergence of the method. Keywords: Gibbs entropy; Shannon entropy; liquid metal 1. Introduction Let pi be the probability of occurrence of a state i, in thermodynamic equilibrium. The Gibbs’ and Von Neumann’s formulas for the entropy [1,2], S/kB = − ∑ pi ln pi, (1) i are mathematically equivalent to the information measure defined by Shannon [3]. Entropy is thus a statistical quantity that can be calculated without reference to the underlying energetics that created the probability distribution, as recognized by Jaynes [4]. Previously we applied this concept to calculate the entropy of liquid aluminum, copper and a liquid aluminum-copper alloy binary alloy [5], using densities and correlation functions obtained from first principles molecular dynamics simulations that are nominally exact within the approximations of electronic density functional theory. -

Theoretical Confirmation of Two High-Pressure Rhombohedral Phase
Stability of rhombohedral phases in vanadium at high-pressure and high-temperature: first-principles investigations Yi X. Wang,1,2 Q. Wu,1 Xiang R. Chen,2,* and Hua Y. Geng1,* Abstract: The pressure-induced transition of vanadium from BCC to rhombohedral structures is unique and intriguing among transition metals. In this work, the stability of these phases is revisited by using density functional theory. At finite temperatures, a novel transition of rhombohedral phases back to BCC phase induced by thermal electrons is discovered. This reentrant transition is found not driven by phonons, instead it is the electronic entropy that stabilizes the latter phase, which is totally out of expectation. Parallel to this transition, we find a peculiar and strong increase of the shear modulus C44 with increasing temperature. It is counter-intuitive in the sense that it suggests an unusual harding mechanism of vanadium by temperature. With these stability analyses, the high-pressure and finite-temperature phase diagram of vanadium is proposed. Furthermore, the dependence of the stability of RH phases on the Fermi energy and chemical environment is investigated. The results demonstrate that the position of the Fermi level has a significant impact on the phase stability, and follows the band-filling argument. Besides the Fermi surface nesting, we find that the localization/delocalization of the d orbitals also contributes to the instability of rhombohedral distortions in vanadium. 1National Key Laboratory of Shock Wave and Detonation Physics, Institute of Fluid Physics, CAEP; P.O. Box 919-102, Mianyang 621900, Sichuan, People’s Republic of China. 2College of Physical Science and Technology, Sichuan University, Chengdu 610064, China. -

A Thermodynamic Basis for the Electronic Properties of Molten Semiconductors: the Role of Electronic Entropy
A thermodynamic basis for the electronic properties of molten semiconductors: the role of electronic entropy The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Rinzler, Charles C., and A. Allanore. “A Thermodynamic Basis for the Electronic Properties of Molten Semiconductors: The Role of Electronic Entropy.” Philosophical Magazine 97, 8 (December 2016): 561–571 © 2016 Informa UK limited, trading as Taylor & Francis group As Published https://doi.org/10.1080/14786435.2016.1269968 Publisher Taylor & Francis Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/114781 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ A Thermodynamic Basis for the Electronic Properties of Molten Semiconductors: The Role of Electronic Entropy Charles C. Rinzler2 , A. Allanore1 1Massachusetts Institute of Technology, Department of Materials Science and Engineering 77 Massachusetts Avenue, Room 13-5066, Cambridge, MA, USA 02139 e-mail address: [email protected] 617-452-2758 2 e-mail address: [email protected] 617-314-1999 1 A Thermodynamic Basis for the Electronic Properties of Molten Semiconductors: The Role of Electronic Entropy The thermodynamic origin of a relation between features of the phase diagrams and the electronic properties of molten semiconductors is provided. Leveraging a quantitative connection between electronic properties and entropy, a criterion is derived to establish whether a system will retain its semiconducting properties in the molten phase. It is shown that electronic entropy is critical to the thermodynamics of molten semiconductor systems, driving key features of phase diagrams including, for example, miscibility gaps. -

Temperature & Excitations
Temperature & Excitations Lecture 10 3/19/18 Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky 1 So far we worked on the ground state (T=0) What happens at finite temperature? When are temperature effects are important for your problem? When are zero-temperature calculations relevant? How to use T=0 calculations to get T-dependent properties? Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky 2 Above the ground state Quantum Mechanics (DFT) gives you the ground state energy When temperature increases, energy increases Additional energy is contained in excitations Which excitations are relevant for your property? Average energy above ground state is a measure of temperature Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky 3 Changes in Materials with Temperature Crystal structure, phase changes Surface structure, concentration profile Chemical composition Electrical and thermal conductivity Bulk modulus Volume Different approaches may be needed for each of these Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky 4 Types of excitations Electronic Magnetic Vibrational Configurational These can couple and take exotic shapes Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky 5 What is temperature? Temperature is a parameter that is equal for two different systems when they are in thermal equilibrium T is a measure of the mean of the energy of the translational, vibrational and rotational (classical) motions of atoms/molecules from the CompassMR simulator from -

Methods for Large-Scale Density Functional Calculations on Metallic Systems Jolyon Aarons, Misbah Sarwar, David Thompsett, and Chris-Kriton Skylaris
Perspective: Methods for large-scale density functional calculations on metallic systems Jolyon Aarons, Misbah Sarwar, David Thompsett, and Chris-Kriton Skylaris Citation: J. Chem. Phys. 145, 220901 (2016); doi: 10.1063/1.4972007 View online: http://dx.doi.org/10.1063/1.4972007 View Table of Contents: http://aip.scitation.org/toc/jcp/145/22 Published by the American Institute of Physics THE JOURNAL OF CHEMICAL PHYSICS 145, 220901 (2016) Perspective: Methods for large-scale density functional calculations on metallic systems Jolyon Aarons,1 Misbah Sarwar,2 David Thompsett,2 and Chris-Kriton Skylaris1,a) 1School of Chemistry, University of Southampton, Southampton SO17 1BJ, United Kingdom 2Johnson Matthey Technology Centre, Sonning Common, Reading, United Kingdom (Received 14 July 2016; accepted 28 November 2016; published online 15 December 2016) Current research challenges in areas such as energy and bioscience have created a strong need for Density Functional Theory (DFT) calculations on metallic nanostructures of hundreds to thousands of atoms to provide understanding at the atomic level in technologically important processes such as catalysis and magnetic materials. Linear-scaling DFT methods for calculations with thousands of atoms on insulators are now reaching a level of maturity. However such methods are not applicable to metals, where the continuum of states through the chemical potential and their partial occupancies provide significant hurdles which have yet to be fully overcome. Within this perspective we outline the theory of DFT calculations on metallic systems with a focus on methods for large-scale calculations, as required for the study of metallic nanoparticles. We present early approaches for electronic energy minimization in metallic systems as well as approaches which can impose partial state occupancies from a thermal distribution without access to the electronic Hamiltonian eigenvalues, such as the classes of Fermi operator expansions and integral expansions.