Minding the Gaps to Promote Thrombus Growth and Stability
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

ORIGINAL ARTICLE Flow Cytometric Protein Expression Profiling As a Systematic Approach for Developing Disease-Specific Assays
Leukemia (2006) 20, 2102–2110 & 2006 Nature Publishing Group All rights reserved 0887-6924/06 $30.00 www.nature.com/leu ORIGINAL ARTICLE Flow cytometric protein expression profiling as a systematic approach for developing disease-specific assays: identification of a chronic lymphocytic leukaemia-specific assay for use in rituximab-containing regimens AC Rawstron, R de Tute, AS Jack and P Hillmen Haematological Malignancy Diagnostic Service (HMDS), Leeds Teaching Hospitals, Leeds, UK Depletion of disease below the levels detected by sensitive sustained remissions only occur in patients achieving an MRD- minimal residual disease (MRD) assays is associated with negative complete response.12 Therefore MRD is increasingly prolonged survival in chronic lymphocytic leukaemia (CLL). being used as an end point for therapeutic trials, and several Flow cytometric MRD assays are now sufficiently sensitive and rapid to guide the duration of therapy in CLL, but generally rely studies are now using the assessment of MRD to define the on assessment of CD20 expression, which cannot be accurately duration of therapy. measured during and after therapeutic approaches containing Approaches using allele-specific oligonucleotide polymerase rituximab. The aim of this study was to use analytical software chain reaction (ASO-PCR) to the immunoglobulin gene of the developed for microarray analysis to provide a systematic B-CLL cell are generally accepted to show the highest sensitivity approach for MRD flow assay development. Samples from CLL for MRD detection. However, more recent four-colour ap- patients (n ¼ 49), normal controls (n ¼ 21) and other B-lympho- proaches show sensitivities nearing that of ASO-PCR6,11,13 with proliferative disorders (n ¼ 12) were assessed with a panel of 66 antibodies. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Flow Reagents Single Color Antibodies CD Chart
CD CHART CD N° Alternative Name CD N° Alternative Name CD N° Alternative Name Beckman Coulter Clone Beckman Coulter Clone Beckman Coulter Clone T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells CD1a T6, R4, HTA1 Act p n n p n n S l CD99 MIC2 gene product, E2 p p p CD223 LAG-3 (Lymphocyte activation gene 3) Act n Act p n CD1b R1 Act p n n p n n S CD99R restricted CD99 p p CD224 GGT (γ-glutamyl transferase) p p p p p p CD1c R7, M241 Act S n n p n n S l CD100 SEMA4D (semaphorin 4D) p Low p p p n n CD225 Leu13, interferon induced transmembrane protein 1 (IFITM1). p p p p p CD1d R3 Act S n n Low n n S Intest CD101 V7, P126 Act n p n p n n p CD226 DNAM-1, PTA-1 Act n Act Act Act n p n CD1e R2 n n n n S CD102 ICAM-2 (intercellular adhesion molecule-2) p p n p Folli p CD227 MUC1, mucin 1, episialin, PUM, PEM, EMA, DF3, H23 Act p CD2 T11; Tp50; sheep red blood cell (SRBC) receptor; LFA-2 p S n p n n l CD103 HML-1 (human mucosal lymphocytes antigen 1), integrin aE chain S n n n n n n n l CD228 Melanotransferrin (MT), p97 p p CD3 T3, CD3 complex p n n n n n n n n n l CD104 integrin b4 chain; TSP-1180 n n n n n n n p p CD229 Ly9, T-lymphocyte surface antigen p p n p n -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -
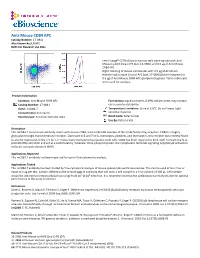
Anti-Mouse CD84 APC Catalog Number: 17‐0841 Also Known As:SLAMF5 RUO: for Research Use Only
Anti-Mouse CD84 APC Catalog Number: 17‐0841 Also Known As:SLAMF5 RUO: For Research Use Only Left: Lineagelo C57BL/6 bone marrow cells were stained with Anti‐ Mouse Ly‐6A/E (Sca‐1) PE (cat. 12‐5981) and 0.5 µg of Anti‐Mouse CD84 APC. Right: Staining of mouse splenocytes with 0.5 µg of Armenian Hamster IgG Isotype Control APC (cat. 17‐4888) (blue histogram) or 0.5 µg of Anti‐Mouse CD84 APC (purple histogram). Total viable cells were used for analysis. Product Information Contents: Anti‐Mouse CD84 APC Formulation: aqueous buffer, 0.09% sodium azide, may contain Catalog Number: 17‐0841 carrier protein/stabilizer Clone: mCD84.7 Temperature Limitation: Store at 2‐8°C. Do not freeze. Light Concentration: 0.2 mg/mL sensitive material. Host/Isotype: Armenian Hamster IgG1 Batch Code: Refer to Vial Use By: Refer to Vial Description This mCD84.7 monoclonal antibody reacts with mouse CD84, a 64‐ to 82‐kDa member of the SLAM family of Ig receptors. CD84 is a highly glycosylated single transmembrane receptor. Expressed in B and T cells, monocytes, platelets, and thymocytes, this receptor was recently found to also be expressed on Sca‐1+c‐kit+Lin‐ mouse bone marrow hematopoietic stem cells. CD84 has been reported to bind itself homophilically to promote IFNγ secretion and act as a costimulatory molecule. Once phosphorylated, the cytoplasmic tail binds signaling lymphocyte activation molecule‐associated protein (SAP). Applications Reported This mCD84.7 antibody has been reported for use in flow cytometric analysis. Applications Tested This mCD84.7 antibody has been tested by flow cytometric analysis of mouse splenocytes and bone marrow. -

Expression of Inflammation-Related Intercellular Adhesion Molecules in Cardiomyocytes in Vitro and Modulation by Pro-Inflammatory Agents
in vivo 30: 213-218 (2016) Expression of Inflammation-related Intercellular Adhesion Molecules in Cardiomyocytes In Vitro and Modulation by Pro-inflammatory Agents IBRAHIM EL-BATTRAWY1,2,3, EROL TÜLÜMEN2,3, SIEGFRIED LANG1,2,3, IBRAHIM AKIN2,3, MICHAEL BEHNES2,3, XIABO ZHOU1,2,3,4, MARTIN MAVANY2, PETER BUGERT5, KAREN BIEBACK5, MARTIN BORGGREFE2,3 and ELIF ELMAS2,3 1Division of Experimental Cardiology and 2First Department of Medicine, Medical Faculty Mannheim, University Heidelberg, Mannheim, Germany; 3German Center for Cardiovascular Research, Partner Site, Heidelberg-Mannheim, Mannheim, Germany; 4Institute of Cardiovascular Research, Sichuan Medical University, Luzhou, Sichuan, P.R. China; 5Institute for Transfusion Medicine and Immunology, Mannheim, Germany Abstract. Background: Cell-surface adhesion molecules not result in increased levels of CD31 (p>0.10). The pro- regulate multiple intercellular and intracellular processes and inflammatory agents LPS and thrombin had no effect on the play important roles in inflammation by facilitating leukocyte expression of MADCAM1 and F11R. Conclusion: endothelial transmigration. Whether cardiomyocytes express Inflammation-related cell-adhesion molecules CD31, surface-adhesion molecules related to inflammation and the MADCAM1 and F11R were shown to be expressed on the effect of pro-inflammatory mediators remain unknown. surface of human cardiomyocytes in an in vitro model. Materials and Methods: In the present study, the expression Incubation with LPS or thrombin resulted in increased of different cell-adhesion molecules (CD11a, CD11b, CD31, expression of CD31, however, it did not modify the expression CD62P, CD162, F11 receptor and mucosal vascular addressin of the cell adhesion molecules MADCAM1 and F11R. cell adhesion molecule 1 (MADCAM1)) and the effect of pro- inflammatory mediators were investigated in an in vitro model Coronary artery disease remains the leading cause of mortality of human cardiomyocytes. -

Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis
biomedicines Review Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis Thijs J. Sluiter 1,2 , Jaap D. van Buul 3 , Stephan Huveneers 4, Paul H. A. Quax 1,2 and Margreet R. de Vries 1,2,* 1 Department of Vascular Surgery, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands; [email protected] (T.J.S.); [email protected] (P.H.A.Q.) 2 Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands 3 Sanquin Research and Landsteiner Laboratory, Leeuwenhoek Centre for Advanced Microscopy, Swammerdam Institute for Life Sciences, University of Amsterdam, 1066 CX Amsterdam, The Netherlands; [email protected] 4 Department of Medical Biochemistry, Amsterdam Cardiovascular Sciences, Amsterdam University Medical Center, Location AMC, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-(71)-526-5147 Abstract: The vascular endothelium is a highly specialized barrier that controls passage of fluids and migration of cells from the lumen into the vessel wall. Endothelial cells assist leukocytes to extravasate and despite the variety in the specific mechanisms utilized by different leukocytes to cross different vascular beds, there is a general principle of capture, rolling, slow rolling, arrest, crawling, and ultimately diapedesis via a paracellular or transcellular route. In atherosclerosis, the barrier function of the endothelium is impaired leading to uncontrolled leukocyte extravasation and Citation: Sluiter, T.J.; van Buul, J.D.; vascular leakage. This is also observed in the neovessels that grow into the atherosclerotic plaque Huveneers, S.; Quax, P.H.A.; de Vries, leading to intraplaque hemorrhage and plaque destabilization. -

Signalling Lymphocyte Activation Molecule Family Member 9 Is Found on Select Subsets of Antigen-Presenting Cells and Promotes Resistance to Salmonella Infection
IMMUNOLOGY ORIGINAL ARTICLE Signalling lymphocyte activation molecule family member 9 is found on select subsets of antigen-presenting cells and promotes resistance to Salmonella infection Timothy J. Wilson,1,2 Summary Simon Clare,3 Joseph Mikulin,1 Signalling lymphocyte activation molecule family member 9 (SLAMF9) is Christopher M. Johnson,4 an orphan receptor of the CD2/SLAM family of leucocyte surface pro- Katherine Harcourt,3 Paul A. teins. Examination of SLAMF9 expression and function indicates that Lyons,2,5 Gordon Dougan3,5 and SLAMF9 promotes inflammation by specialized subsets of antigen-pre- Kenneth G. C. Smith2,5 1 senting cells. Within healthy liver and circulating mouse peripheral blood Department of Microbiology, Miami + À low 2 mononuclear cells, SLAMF9 is expressed on CD11b , Ly6C , CD11c , University, Oxford, OH, USA, Department low + + of Medicine, Cambridge Institute for Medical F4/80 , MHC-II ,CX3CR1 mononuclear phagocytes as well as plasma- Research, University of Cambridge, cytoid dendritic cells. In addition, SLAMF9 can be found on peritoneal Cambridge, 3Wellcome Trust Sanger Institute, B1 cells and small (F4/80low), but not large (F4/80high), peritoneal macro- 4 Hinxton, Cambridge, MRC Laboratory of phages. Upon systemic challenge with Salmonella enterica Typhimurium, À/À Molecular Biology, Cambridge, and Slamf9 mice were impaired in their ability to clear the infection from 5Cambridge Institute for Therapeutic Immunology and Infectious Disease, the liver. In humans, SLAMF9 is up-regulated upon differentiation of Department of Medicine, University of monocytes into macrophages, and lipopolysaccharide stimulation of Cambridge, Cambridge, UK PMA-differentiated, SLAMF9 knockdown THP-1 cells showed an essential role of SLAMF9 in production of granulocyte–macrophage colony-stimu- doi:10.1111/imm.13169 lating factor, tumour necrosis factor-a, and interleukin-1b. -

The VE-Cadherin/Amotl2 Mechanosensory Pathway Suppresses Aortic In�Ammation and the Formation of Abdominal Aortic Aneurysms
The VE-cadherin/AmotL2 mechanosensory pathway suppresses aortic inammation and the formation of abdominal aortic aneurysms Yuanyuan Zhang Karolinska Institute Evelyn Hutterer Karolinska Institute Sara Hultin Karolinska Institute Otto Bergman Karolinska Institute Maria Forteza Karolinska Institute Zorana Andonovic Karolinska Institute Daniel Ketelhuth Karolinska University Hospital, Stockholm, Sweden Joy Roy Karolinska Institute Per Eriksson Karolinska Institute Lars Holmgren ( [email protected] ) Karolinska Institute Article Keywords: arterial endothelial cells (ECs), vascular disease, abdominal aortic aneurysms Posted Date: June 15th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-600069/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License The VE-cadherin/AmotL2 mechanosensory pathway suppresses aortic inflammation and the formation of abdominal aortic aneurysms Yuanyuan Zhang1, Evelyn Hutterer1, Sara Hultin1, Otto Bergman2, Maria J. Forteza2, Zorana Andonovic1, Daniel F.J. Ketelhuth2,3, Joy Roy4, Per Eriksson2 and Lars Holmgren1*. 1Department of Oncology-Pathology, BioClinicum, Karolinska Institutet, Stockholm, Sweden. 2Department of Medicine Solna, BioClinicum, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden. 3Department of Cardiovascular and Renal Research, Institutet of Molecular Medicine, Univ. of Southern Denmark, Odense, Denmark 4Department of Molecular Medicine and Surgery, Karolinska Institutet, Karolinska University Hospital, Stockholm,