Direct Reprogramming of Fibroblasts Into Muscle Or Neural Lineages by Using Single Transcription Factor with Or Without Myod Transactivation Domain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The REST/NRSF Pathway As a Central Mechanism in CNS Dysfunction
The REST/NRSF Pathway as a Central Mechanism in CNS Dysfunction Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Alix Warburton April 2015 Disclaimer The data in this thesis is a result of my own work. The material collected for this thesis has not been presented, nor is currently being presented, either wholly or in part for any other degree or other qualification. All of the research, unless otherwise stated, was performed in the Department of Physiology and Department of Pharmacology, Institute of Translational Medicine, University of Liverpool. All other parties involved in the research presented here, and the nature of their contribution, are listed in the Acknowledgements section of this thesis. i Acknowledgements First and foremost, I would like to express my upmost gratitude to my primary and secondary supervisors Professor John Quinn (a.k.a Prof. Quinny) and Dr Jill Bubb for all of their support, guidance, wisdom (thank you Jill) and encouragement throughout my PhD; I could not have wished for a better pair. I am also extremely grateful to the BBSRC for funding my PhD project. I would also like to extend my thanks to Dr Graeme Sills for providing samples and assistance with my work on the SANAD epilepsy project, Dr Fabio Miyajima for offering his knowledge and knowhow on many occasions, Dr Gerome Breen for being a bioinformatics wizard and providing support on several projects, Dr Minyan Wang’s lab for their help and hospitality during my 3 month visit to Xi'an Jiaotong-Liverpool University, Dr Roshan Koron for assisting with the breast cancer study, Dr Chris Murgatroyd for his invaluable advice on ChIP and Professor Dan Rujescu’s lab for providing clinical samples and support with statistical analyses on the schizophrenia project. -

Intrinsic Specificity of DNA Binding and Function of Class II Bhlh
INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS APPROVED BY SUPERVISORY COMMITTEE Jane E. Johnson Ph.D. Helmut Kramer Ph.D. Genevieve Konopka Ph.D. Raymond MacDonald Ph.D. INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS by BRADFORD HARRIS CASEY DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas December, 2016 DEDICATION This work is dedicated to my family, who have taught me pursue truth in all forms. To my grandparents for inspiring my curiosity, my parents for teaching me the value of a life in the service of others, my sisters for reminding me of the importance of patience, and to Rachel, who is both “the beautiful one”, and “the smart one”, and insists that I am clever and beautiful, too. Copyright by Bradford Harris Casey, 2016 All Rights Reserved INTRINSIC SPECIFICITY OF BINDING AND REGULATORY FUNCTION OF CLASS II BHLH TRANSCRIPTION FACTORS Publication No. Bradford Harris Casey The University of Texas Southwestern Medical Center at Dallas, 2016 Jane E. Johnson, Ph.D. PREFACE Embryonic development begins with a single cell, and gives rise to the many diverse cells which comprise the complex structures of the adult animal. Distinct cell fates require precise regulation to develop and maintain their functional characteristics. Transcription factors provide a mechanism to select tissue-specific programs of gene expression from the shared genome. -

Download and Analyze Large-Scale Cancer Studies Such As the Cancer Genome Atlas (TCGA) Studies for Different Cancers (189)
MECHANISMS OF TREATMENT INDUCED CELL PLASTICITY IN PROSTATE CANCER by Daksh Thaper BSc, Simon Fraser University, 2012 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Doctor of Philosophy in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) August 2018 © Daksh Thaper, 2018 i Examining Committee The following individuals certify that they have read, and recommend to the Faculty of Graduate and Postdoctoral Studies for acceptance, the dissertation entitled: Mechanism of treatment induced cell plasticity in prostate cancer in partial fulfillment of the requirements submitted by Daksh Thaper for the degree of Doctor of Philosophy in Experimental Medicine Examining Committee: Dr. Amina Zoubeidi, Associate Professor, Department of Urologic Sciences Supervisor Dr. Michael Cox, Associate Professor, Department of Urologic Sciences Supervisory Committee Member Dr. Christopher Maxwell, Associate Professor, Department of Pediatrics Supervisory Committee Member Dr. Shoukat Dedhar, Professor, Department of Biochemistry Supervisory Committee Member Dr. Calvin Roskelley, Professor, Department of Cellular and Physiological Sciences University Examiner Dr. Wan Lam, Professor, Department of Pathology and Laboratory Medicine University Examiner ii Abstract While effective, resistance to 1st generation and 2nd generation androgen receptor (AR) pathway inhibitors is inevitable, creating a need to study the mechanisms by which prostate cancer (PCa) cells become resistant to these treatments. At its core, resistance can be categorized into AR driven and non-AR driven. The research presented in this thesis explores the hypothesis that non-AR driven resistance mechanisms exploit cellular plasticity to gain a survival advantage. Previous research has demonstrated that in response to 1st generation anti-androgens, epithelial PCa cells can undergo an epithelial-mesenchymal transition (EMT); a process implicated in metastatic dissemination of cancer cells throughout the body. -

Integrative Analysis of Single-Cell Genomics Data by Coupled Nonnegative Matrix Factorizations
Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations Zhana Durena,b,1, Xi Chena,b,1, Mahdi Zamanighomia,b,c,1, Wanwen Zenga,b,d, Ansuman T. Satpathyc, Howard Y. Changc, Yong Wange,f, and Wing Hung Wonga,b,c,2 aDepartment of Statistics, Stanford University, Stanford, CA 94305; bDepartment of Biomedical Data Science, Stanford University, Stanford, CA 94305; cCenter for Personal Dynamic Regulomes, Stanford University, Stanford, CA 94305; dMinistry of Education Key Laboratory of Bioinformatics, Bioinformatics Division and Center for Synthetic & Systems Biology, Department of Automation, Tsinghua University, 100084 Beijing, China; eAcademy of Mathematics and Systems Science, Chinese Academy of Sciences, 100080 Beijing, China; and fCenter for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, 650223 Kunming, China Contributed by Wing Hung Wong, June 14, 2018 (sent for review April 4, 2018; reviewed by Andrew D. Smith and Nancy R. Zhang) When different types of functional genomics data are generated on Approach single cells from different samples of cells from the same hetero- Coupled NMF. We first introduce our approach in general terms. geneous population, the clustering of cells in the different samples Let O be a p1 by n1 matrix representing data on p1 features for n1 should be coupled. We formulate this “coupled clustering” problem units in the first sample; then a “soft” clustering of the units in as an optimization problem and propose the method of coupled this sample can be obtained from a nonnegative factorization nonnegative matrix factorizations (coupled NMF) for its solution. O = W1H1 as follows: The ith column of W1 gives the mean The method is illustrated by the integrative analysis of single-cell vector for the ith cluster of units, while the jth column of H1 gives RNA-sequencing (RNA-seq) and single-cell ATAC-sequencing (ATAC- the assignment weights of the jth unit to the different clusters. -

Investigation of Differentially Expressed Genes in Nasopharyngeal Carcinoma by Integrated Bioinformatics Analysis
916 ONCOLOGY LETTERS 18: 916-926, 2019 Investigation of differentially expressed genes in nasopharyngeal carcinoma by integrated bioinformatics analysis ZhENNING ZOU1*, SIYUAN GAN1*, ShUGUANG LIU2, RUjIA LI1 and jIAN hUANG1 1Department of Pathology, Guangdong Medical University, Zhanjiang, Guangdong 524023; 2Department of Pathology, The Eighth Affiliated hospital of Sun Yat‑sen University, Shenzhen, Guangdong 518033, P.R. China Received October 9, 2018; Accepted April 10, 2019 DOI: 10.3892/ol.2019.10382 Abstract. Nasopharyngeal carcinoma (NPC) is a common topoisomerase 2α and TPX2 microtubule nucleation factor), malignancy of the head and neck. The aim of the present study 8 modules, and 14 TFs were identified. Modules analysis was to conduct an integrated bioinformatics analysis of differ- revealed that cyclin-dependent kinase 1 and exportin 1 were entially expressed genes (DEGs) and to explore the molecular involved in the pathway of Epstein‑Barr virus infection. In mechanisms of NPC. Two profiling datasets, GSE12452 and summary, the hub genes, key modules and TFs identified in GSE34573, were downloaded from the Gene Expression this study may promote our understanding of the pathogenesis Omnibus database and included 44 NPC specimens and of NPC and require further in-depth investigation. 13 normal nasopharyngeal tissues. R software was used to identify the DEGs between NPC and normal nasopharyngeal Introduction tissues. Distributions of DEGs in chromosomes were explored based on the annotation file and the CYTOBAND database Nasopharyngeal carcinoma (NPC) is a common malignancy of DAVID. Gene ontology (GO) and Kyoto Encyclopedia of occurring in the head and neck. It is prevalent in the eastern Genes and Genomes (KEGG) pathway enrichment analysis and southeastern parts of Asia, especially in southern China, were applied. -

Transcriptomic and Epigenomic Characterization of the Developing Bat Wing
ARTICLES OPEN Transcriptomic and epigenomic characterization of the developing bat wing Walter L Eckalbar1,2,9, Stephen A Schlebusch3,9, Mandy K Mason3, Zoe Gill3, Ash V Parker3, Betty M Booker1,2, Sierra Nishizaki1,2, Christiane Muswamba-Nday3, Elizabeth Terhune4,5, Kimberly A Nevonen4, Nadja Makki1,2, Tara Friedrich2,6, Julia E VanderMeer1,2, Katherine S Pollard2,6,7, Lucia Carbone4,8, Jeff D Wall2,7, Nicola Illing3 & Nadav Ahituv1,2 Bats are the only mammals capable of powered flight, but little is known about the genetic determinants that shape their wings. Here we generated a genome for Miniopterus natalensis and performed RNA-seq and ChIP-seq (H3K27ac and H3K27me3) analyses on its developing forelimb and hindlimb autopods at sequential embryonic stages to decipher the molecular events that underlie bat wing development. Over 7,000 genes and several long noncoding RNAs, including Tbx5-as1 and Hottip, were differentially expressed between forelimb and hindlimb, and across different stages. ChIP-seq analysis identified thousands of regions that are differentially modified in forelimb and hindlimb. Comparative genomics found 2,796 bat-accelerated regions within H3K27ac peaks, several of which cluster near limb-associated genes. Pathway analyses highlighted multiple ribosomal proteins and known limb patterning signaling pathways as differentially regulated and implicated increased forelimb mesenchymal condensation in differential growth. In combination, our work outlines multiple genetic components that likely contribute to bat wing formation, providing insights into this morphological innovation. The order Chiroptera, commonly known as bats, is the only group of To characterize the genetic differences that underlie divergence in mammals to have evolved the capability of flight. -

Dynamic Regulatory Module Networks for Inference of Cell Type
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.18.210328; this version posted July 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Dynamic regulatory module networks for inference of cell type specific transcriptional networks Alireza Fotuhi Siahpirani1,2,+, Deborah Chasman1,8,+, Morten Seirup3,4, Sara Knaack1, Rupa Sridharan1,5, Ron Stewart3, James Thomson3,5,6, and Sushmita Roy1,2,7* 1Wisconsin Institute for Discovery, University of Wisconsin-Madison 2Department of Computer Sciences, University of Wisconsin-Madison 3Morgridge Institute for Research 4Molecular and Environmental Toxicology Program, University of Wisconsin-Madison 5Department of Cell and Regenerative Biology, University of Wisconsin-Madison 6Department of Molecular, Cellular, & Developmental Biology, University of California Santa Barbara 7Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison 8Present address: Division of Reproductive Sciences, Department of Obstetrics and Gynecology, University of Wisconsin-Madison +These authors contributed equally. *To whom correspondence should be addressed. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.18.210328; this version posted July 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Changes in transcriptional regulatory networks can significantly alter cell fate. To gain insight into transcriptional dynamics, several studies have profiled transcriptomes and epigenomes at different stages of a developmental process. -

High-Throughput Biochemical Analysis of In-Vivo Location Data Reveals Novel Distinct Classes of POU5F1(Oct4)/DNA Complexes. ABST
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press High-throughput Biochemical Analysis of in-vivo Location Data Reveals Novel Distinct Classes of POU5F1(Oct4)/DNA complexes. Dean Tantin1^, Matthew Gemberling2^, Catherine Callister1, William Fairbrother* 2,3 1 2 3 Department of Pathology, University of Utah School of Medicine, Salt Lake City, Utah 84112; MCB Department, Brown University, Providence, Rhode Island. Center for Computational Molecular Biology, Brown University, Providence, Rhode Island 02912 ^ These Authors contributed equally to this work. *To whom correspondence should be addressed: Will Fairbrother, Brown University, Providence RI [email protected] ABSTRACT: The transcription factor POU5F1 is a key regulator of embryonic stem (ES) cell pluripotency and a known oncoprotein. We have developed a novel high-throughput binding assay called MEGAshift (microarray evaluation of genomic aptamers by shift) that we use to pinpoint the exact location, affinity and stoichiometry of the DNA-protein complexes identified by chromatin immunoprecipitation studies. We consider all genomic regions identified as POU5F1-ChIP-enriched in both human and mouse. Compared to regions that are ChIP- enriched in a single species, we find these regions more likely to be near actively transcribed genes in ES cells. We re-synthesize these genomic regions as a pool of tiled 35-mers. This oligonucleotide pool is then assayed for binding to recombinant POU5F1 by gel shift. The degree of binding for each oligonucleotide is accurately measured on a specially designed microarray. We explore the relationship between experimentally determined and computationally predicted binding strengths, find many novel functional combinations of POU5F1 half sites and demonstrate efficient motif discovery by incorporating binding information into a motif finding algorithm. -

Novel Mutations Segregating with Complete Androgen Insensitivity Syndrome and Their Molecular Characteristics
International Journal of Molecular Sciences Article Novel Mutations Segregating with Complete Androgen Insensitivity Syndrome and Their Molecular Characteristics Agnieszka Malcher 1, Piotr Jedrzejczak 2, Tomasz Stokowy 3 , Soroosh Monem 1, Karolina Nowicka-Bauer 1, Agnieszka Zimna 1, Adam Czyzyk 4, Marzena Maciejewska-Jeske 4, Blazej Meczekalski 4, Katarzyna Bednarek-Rajewska 5, Aldona Wozniak 5, Natalia Rozwadowska 1 and Maciej Kurpisz 1,* 1 Institute of Human Genetics, Polish Academy of Sciences, 60-479 Poznan, Poland; [email protected] (A.M.); [email protected] (S.M.); [email protected] (K.N.-B.); [email protected] (A.Z.); [email protected] (N.R.) 2 Division of Infertility and Reproductive Endocrinology, Department of Gynecology, Obstetrics and Gynecological Oncology, Poznan University of Medical Sciences, 60-535 Poznan, Poland; [email protected] 3 Department of Clinical Science, University of Bergen, 5020 Bergen, Norway; [email protected] 4 Department of Gynecological Endocrinology, Poznan University of Medical Sciences, 60-535 Poznan, Poland; [email protected] (A.C.); [email protected] (M.M.-J.); [email protected] (B.M.) 5 Department of Clinical Pathology, Poznan University of Medical Sciences, 60-355 Poznan, Poland; [email protected] (K.B.-R.); [email protected] (A.W.) * Correspondence: [email protected]; Tel.: +48-61-6579-202 Received: 11 October 2019; Accepted: 29 October 2019; Published: 30 October 2019 Abstract: We analyzed three cases of Complete Androgen Insensitivity Syndrome (CAIS) and report three hitherto undisclosed causes of the disease. RNA-Seq, Real-timePCR, Western immunoblotting, and immunohistochemistry were performed with the aim of characterizing the disease-causing variants. -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors. -
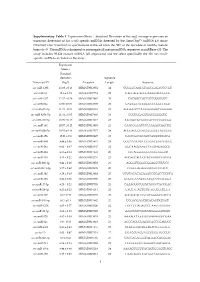
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Supplementary Table S2. List of Genes with Expression That Is Positively Correlated (Pearson Correlation Coefficient P>0.3)
Supplementary Table S2. List of genes with expression that is positively correlated (Pearson correlation coefficient p>0.3) with HOXA9 expression in the study by Sun et al (1). Most HOXA genes (indicated by bold face) show highly significant positive correlation with HOXA9 expression, except for HOXA6 and HOXA13, similar to our findings in the UCSF and MDA tumor sets. Note that HOXA11, which did not demonstrate a statistically significant positive correlation with HOXA9 expression in both the UCSF and MDA tumor sets, demonstrates a substantially lower correlation coefficient relative to the other HOXA genes that positively correlate with HOXA9 expression in the study by Sun et al. These data were obtained from the online ONCOMINE database (www.oncomine.org) (2) searching for transcripts positively correlated with HOXA9 expression. Gene Name Gene Symbol Reporter ID Correlation (p) homeobox A9 HOXA9 214651_s_at .7682 homeobox A9 HOXA9 209905_at .7682 homeobox A10 HOXA10 213150_at .6058 homeobox A7 HOXA7 235753_at .6058 homeobox A10 HOXA10 213147_at .6058 homeobox A7 HOXA7 206847_s_at .6058 homeobox A4 HOXA4 206289_at .5741 homeobox A2 HOXA2 1557051_s_at .5379 homeobox A1 HOXA1 214639_s_at .5379 homeobox A3 HOXA3 235521_at .5379 homeobox B2 HOXB2 205453_at .5379 homeobox B3 HOXB3 228904_at .5379 EST EST 1555907_at .5379 homeobox A4 HOXA4 230080_at .5379 homeobox A2 HOXA2 228642_at .5379 homeobox A2 HOXA2 1557050_at .5379 homeobox A5 HOXA5 213844_at .5379 homeobox A2 HOXA2 214457_at .5113 homeobox B7 HOXB7 204778_x_at .5042 homeobox C6 HOXC6