Winnie Wing Yin Tong
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for High-Grade Anal Intraepithelial Neoplasia
Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for High-Grade Anal Intraepithelial Neoplasia Using The BARRX™ Anorectal Wand (Clinical Protocol 01-2017) AUGUST 31, 2017 Page 1 of 40 Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia PROTOCOL SIGNATURE PAGE I, , Principal Investigator at site , agree to conduct and follow this protocol: PROTOCOL 01-2017: A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for Anal Intraepithelial Neoplasia using the Barrx™ Anorectal Wand as written according to FDA guidelines. I understand that no deviations from the above protocol may be made without written permission from the Protocol Chair(s). _________________________________ _____________________ Signature Date (mm/dd/yyyy) Page 2 of 40 Protocol 01-2017: RFA for Anal Intraepithelial Neoplasia P ROT O COL S U M M ARY Title A Safety and Efficacy Trial of Circumferential Anal Canal Radiofrequency Ablation for Anal Intraepithelial Neoplasia using the Barrx™ Anorectal Wand Design Multi-center prospective trial involving up to 70 subjects Subject Population HIV-positive and HIV-negative subjects with intra-anal intraepithelial neoplasia (AIN) containing at least one high- grade squamous intraepithelial lesions (HSIL) involving the squamocolumnar junction (SCJ). Objective Assess the safety, and efficacy of circumferential radiofrequency ablation (RFA) to the anal canal using the FDA cleared Barrx™ Anorectal Wand to eradicate anal -
Detail Report
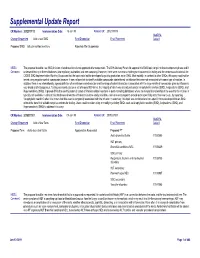
Supplemental Update Report CR Number: 2012319113 Implementation Date: 16-Jan-19 Related CR: 2012319113 MedDRA Change Requested Add a new SMQ Final Disposition Final Placement Code # Proposed SMQ Infusion related reactions Rejected After Suspension MSSO The proposal to add a new SMQ Infusion related reactions is not approved after suspension. The ICH Advisory Panel did approve this SMQ topic to go into the development phase and it Comment: underwent testing in three databases (two regulatory authorities and one company). However, there were numerous challenges encountered in testing and the consensus decision of the CIOMS SMQ Implementation Working Group was that the topic could not be developed to go into production as an SMQ. Most notably, in contrast to other SMQs, this query could not be tested using negative control compounds because it was not possible to identify suitable compounds administered via infusion that were not associated with some type of reaction. In addition, there is no internationally agreed definition of an infusion related reaction and the range of potential reactions associated with the large variety of compounds given by infusion is very broad and heterogenous. Testing was conducted on a set of around 500 terms, the majority of which was already included in Anaphylactic reaction (SMQ), Angioedema (SMQ), and Hypersensitivity (SMQ). It proved difficult to identify potential cases of infusion related reactions in post-marketing databases where the temporal relationship of the event to the infusion is typically not available. In clinical trial databases where this information is more easily available, users are encouraged to provide more specificity about the event, e.g., by reporting “Anaphylactic reaction” when it is known that this event is temporally associated with the infusion. -

Infectious Gastroenteritis Generalised Anxiety Disorder HIV Smoking Associated Cancers
BEST PRACTICE 25 DECEMBER 2009 Infectious gastroenteritis Generalised anxiety disorder HIV bpac nz Smoking associated cancers better medicin e Editorial Team Tony Fraser Professor Murray Tilyard We would like to acknowledge the following people for Clinical Advisory Group their guidance and expertise in developing this edition: Michele Cray Dr Shaun Costello, Dunedin Serena Curtis-Lemuelu Dr Edward Coughlan, Christchurch Dr Rosemary Ikram Professor Tony Dowell, Wellington Dr Cam Kyle Dr Rosemary Ikram, Christchurch Dr Chris Leathart Mr William Pearce, Christchurch Dr Lynn McBain Dr Alan Pithie, Christchurch Adam McRae Dr Gabrielle Ruben, Wellington Janet Maloney-Moni Assoc. Professor Mark Thomas, Auckland Dr Peter Moodie Dr Robyn Toomath, Wellington Associate Professor Jim Reid Dr Neil Whittaker, GP Reviewer, Nelson Associate Professor David Reith Assoc. Professor Michael Williams, Dunedin Professor Murray Tilyard Programme Development Team Rachael Clarke Peter Ellison Best Practice Journal (BPJ) Rebecca Harris Julie Knight ISSN 1177-5645 Noni Richards BPJ, Issue 25, December 2009 Dr Tom Swire Dr AnneMarie Tangney nz Dr Sharyn Willis BPJ is published and owned by bpac Ltd Dave Woods Level 8, 10 George Street, Dunedin, New Zealand. Report Development Team Bpacnz Ltd is an independent organisation that promotes health Justine Broadley care interventions which meet patients’ needs and are evidence Todd Gillies based, cost effective and suitable for the New Zealand context. Lana Johnson We develop and distribute evidence based resources which describe, facilitate and help overcome the barriers to best Web practice. Gordon Smith Bpacnz Ltd is currently funded through contracts with PHARMAC Design and DHBNZ. Michael Crawford Bpacnz Ltd has five shareholders: Procare Health, South Link Management and Administration Health, IPAC, the University of Otago and Pegasus Health. -

Pathology of Anal Cancer
Pathology of Anal Cancer a b Paulo M. Hoff, MD, PhD , Renata Coudry, MD, PhD , a, Camila Motta Venchiarutti Moniz, MD * KEYWORDS Anal cancer Anal squamous intraepithelial neoplasia Squamous cell carcinoma Human papilloma virus (HPV) Molecular KEY POINTS Anal cancer is an uncommon tumor, squamous cell carcinoma (SCC) being the most frequent histology corresponding to 80% of all cases. Human papilloma virus (HPV) infection plays a key role in anal cancer development, en- coding at least three oncoproteins with stimulatory properties. SCC expresses CK5/6, CK 13/19, and p63. P16 is a surrogate marker for the presence of HPV genome in tumor cells. INTRODUCTION Anal cancer accounts for approximately 2.4% of gastrointestinal malignancies.1 Although anal cancer is a rare tumor, its frequency is increasing, especially in high- risk groups.2 Tumors in this location are generally classified as anal canal or anal margin. Squamous cell carcinoma (SCC) is the predominant type of tumor and shares many features with cervical cancer. Oncogenic human papilloma virus (HPV) infection plays a major role in both tumors.3 HIV infection is associated with a higher frequency of HPV-associated premalignant lesions and invasive tumors.4 Normal Anatomy of the Anus The anal canal is the terminal part of the large intestine and is slightly longer in male than in female patients. It measures approximately 4 cm and extends from the rectal ampulla (pelvic floor level) to the anal verge, which is defined as the outer open- ing of the gastrointestinal tract. The anal verge is at the level of the squamous- mucocutaneous junction with the perianal skin.5,6 The authors have nothing to disclose. -

HPV, Anal Dysplasia and Anal Cancer
HPV, anal dysplasia FACT and anal cancer SHEET Published 2016 Summary Anal cancer typically develops over a period of years, beginning with a precancerous condition called anal dysplasia. CONTACT US Anal dysplasia occurs when clusters of abnormal cells form by telephone lesions in the mucosa lining of the anal canal (between the 1-800-263-1638 anus and the rectum). The lesions typically form inside the anal 416-203-7122 canal or just outside the anal opening. by fax 416-203-8284 Although there are over 100 different types of the human papillomavirus (HPV), anal dysplasia is usually caused by certain by e-mail strains of HPV which can be transmitted sexually. HPV can shut [email protected] off the proteins that help prevent dysplasia and cancer cells from developing, therefore leading to HPV-associated diseases by mail such as anal dysplasia. 555 Richmond Street West Suite 505, Box 1104 It is difficult to screen for anal dysplasia since the lesions are Toronto ON M5V 3B1 not detectable by routine examinations. As a result, anal dysplasia is often not detected until it has developed into anal cancer, which can be difficult to treat depending on the severity. Specific screening tests can detect dysplasia or precancerous changes. If these precancers are treated, anal cancer may be prevented. Anal cancer is usually treated with radiation and chemotherapy or with surgery. Although anal dysplasia may be treated successfully, individuals with HIV are at increased risk of it recurring and may need to be monitored closely by a trained physician. Consistent condom use reduces, but does not eliminate, the risk of transmitting HPV. -

Incidence of Recurrent High-Grade Anal Dysplasia in HIV-1-Infected Men and Women Following Infrared Coagulation Ablation: a Retrospective Cohort Study
pathogens Article Incidence of Recurrent High-Grade Anal Dysplasia in HIV-1-Infected Men and Women Following Infrared Coagulation Ablation: A Retrospective Cohort Study Javier Corral 1,2,3,*, David Parés 1,2,3, Francesc García-Cuyás 1,2, Boris Revollo 2,4, Ana Chamorro 2,4, Carla Lecumberri 2,5 , Antoni Tarrats 2,5, Eva Castella 6, Marta Piñol 1,2, Bonaventura Clotet 2,3,7, Sebastià Videla 2,8,*,† and Guillem Sirera 2,4,† 1 Department of General Surgery, Hospital Germans Trias i Pujol, Carretera de Canyet, s/n, Badalona, 08916 Barcelona, Spain; [email protected] (D.P.); [email protected] (F.G.-C.); [email protected] (M.P.) 2 Lluita contra la Sida Foundation, Hospital Universitari Germans Trias i Pujol, Carretera de Canyet, s/n, Badalona, 08916 Barcelona, Spain; brevollo@flsida.org (B.R.); achamorro@flsida.org (A.C.); [email protected] (C.L.); [email protected] (A.T.); [email protected] (B.C.); gsirera@flsida.org (G.S.) 3 School of Medicine, Universitat Autónoma de Barcelona (UAB), Edifici M, Av. de Can Domènech, Bellaterra, 08193 Barcelona, Spain 4 HIV Clinical Unit, Department of Medicine, University Hospital Germans Trias i Pujol, Carretera de Canyet, s/n, Badalona, 08916 Barcelona, Spain 5 Department of Obstetrics and Gynaecology, Hospital Germans Trias i Pujol, Carretera de Canyet, s/n, Badalona, 08916 Barcelona, Spain 6 Department of Pathology, Hospital Germans Trias i Pujol, Carretera de Canyet, s/n, Badalona, Citation: Corral, J.; Parés, D.; 08916 Barcelona, Spain; [email protected] García-Cuyás, F.; Revollo, B.; 7 Retrovirology Laboratory IrsiCaixa Foundation, Carretera de Canyet, s/n, Badalona, 08916 Barcelona, Spain Chamorro, A.; Lecumberri, C.; Tarrats, 8 Department of Clinical Pharmacology, Bellvitge University Hospital/IDIBELL/University of Barcelona, A.; Castella, E.; Piñol, M.; Clotet, B.; Hospitalet de Llobregat, Gran Via de les Corts Catalanes 199–203, 08907 Barcelona, Spain et al. -

Use of Human Papillomavirus Ge
Use of human papillomavirus genotyping and biomarkers for targeted screening of anal dysplasia in human immunodeficiency virus-infected patients Clarisse Dupin, Laurent Siproudhis, Sébastien Hénno, Sophie Minjolle, Cédric Arvieux, Pierre Tattevin To cite this version: Clarisse Dupin, Laurent Siproudhis, Sébastien Hénno, Sophie Minjolle, Cédric Arvieux, et al.. Use of human papillomavirus genotyping and biomarkers for targeted screening of anal dysplasia in human immunodeficiency virus-infected patients. Digestive and Liver Disease, WB Saunders, 2015, 47(5), pp.423-428. 10.1016/j.dld.2015.01.150. hal-01147275 HAL Id: hal-01147275 https://hal-univ-rennes1.archives-ouvertes.fr/hal-01147275 Submitted on 22 Oct 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Use of human papillomavirus genotyping and biomarkers for targeted screening of anal dysplasia in human immunodeficiency virus-infected patients Clarisse Dupin,a,b, Laurent Siproudhis,b,c Sébastien Henno,b,d Sophie Minjolle,a,b Cédric Arvieux,b,e Pierre Tattevinb,e Department of Virology, Pontchaillou -

Christopher Mathews, M.D. UCSD Owen Clinic April 13, 2017 Clinical Case
Screening for Anal Cancer and Precursors in HIV Patients Christopher Mathews, M.D. UCSD Owen Clinic April 13, 2017 Clinical Case • 50 year old asymptomatic physician with HIV infection presented for routine care in May 1999 • CD4=350, HIV viral load 35,000 • Physical exam normal except for 3 cm irregular hard anal mass • Biopsy: invasive squamous cell carcinoma 2 Clinical Case -2 • Resection had positive margins • He was treated with radiotherapy and mitomycin C + 5FU • Severe disabling radiation proctitis • Biopsy at end of treatment showed residual tumor • Abdominal perineal resection in 11/99 • Small bowel obstructionileocolic anastasmosis (3/00) • Bilateral hydronephrosis and renal failure • Declined intervention • Viral load <50 prior to withdrawal of therapy 3 Learning Objectives • Review current HPV vaccination guidelines for HIV-infected adults • Review guidelines related to screening for anal cancer and its precursors in HIV-infected patients • Review the evidence base to support screening • Describe screening options and procedures as related to screening goals and screening procedure expertise 4 Polling question 1 • Which groups of HIV-infected patients should be routinely offered HPV vaccination? (select the one best answer) – All HIV-infected adults who have not yet been vaccinated – All HIV-infected males and females ≤ 21 years of age – All HIV-infected males and females ≤ 26 years of age – The HPV vaccines are unlikely to be immunogenic and shouldn’t be given to HIV-infected patients – The HPV vaccines are live attenuated vaccines and shouldn’t be administered to HIV-infected patients 5 Polling question 2 • According to 2016 CDC guidelines, how many HPV vaccinations are recommended to complete the series for HIV-infected patients? – 1 – 2 – 3 6 HPV VACCINATION UPDATE 7 HPV Vaccination Recommendations: 2016 update 1.Meites E, et al. -

STI / HPV / Cancer
HIV, cancer, HPV & STI’s S. De Wit St Pierre University hospital Brussels HIV and cancer • AIDS-defining malignancies: – Kaposi’s sarcoma HHV8 – Non Hodgkin lymphoma 1985 EBV – Cervical cancer 1993 HPV • Non AIDS-defining malignancies (NADM) is increasing • Linked with virus HPV (Anal), HBV and HCV (Liver), EBV (HL) • Linked with previous immunodeficiency and other factors Background • Before introduction of HAART, ADCs common, including Kaposi’s sarcoma, NHL, and invasive cervical carcinoma • Rate of ADCs significantly increased from early to late pre-HAART era and then significantly decreased following introduction of HAART • Rates of nADCs stable during pre-HAART eras and then significantly increased following introduction of HAART Crum-Cianflone N, et al. AIDS. 2009;23:41-50. AIDS-Defining Cancer SIR = Standardised Incidence Ratio = Nb cases of cancer in the HIV population Expected nb of cases in the general population, calculated with local cancer registry incidence Cancer Incidence in AIDS Patients Cancer type No. cases SIR 95% CI AIDS-defining cancers Kaposi sarcoma 3136 5321 5137 - 5511 • Study of cancer risk in AIDS Non-Hodgkin 3345 32 31 - 33 patients from 1980-2006 lymphoma (N=372,364) Cervical cancer 101 5.6 5.5 - 6.8 • Predominantly male (79%), Non-AIDS-defining cancers non-hispanic black (42%), MSM (42%) Anal cancer 219 27 24 - 31 • Median age of 36 years Liver cancer 86 3.7 3.0 - 4.6 at the onset of AIDS Lung cancer 531 3.0 2.8 - 3.3 Hodgkin lymphoma 184 9.1 7.7 - 11 All non-AIDS 2155 1.7 1.5 - 1.8 related cancers SIR=Standardized Incidence Ratios Simard E, et al. -

Abstracts of the 105Th Annual Congress of the Swiss Society of Surgery, Held in Basel, Switzerland, 16 May – 18 May 2018
May 2018 • Volume 105 • Supplement 3 @BJSurgery Abstracts of the 105th Annual Congress of the Swiss Society of Surgery, held in Basel, Switzerland, 16 May – 18 May 2018 www.bjs.co.uk BJS May 2018 Volume 105 Supplement 3 Abstracts of the 105th Annual Congress of the Swiss Society of Surgery, held in Basel, Switzerland, 16 May–18 May 2018 Disclaimer This abstract book has been produced using author-supplied copy. Editing has been restricted to some corrections of spelling and style where appropriate. The publisher assumes no responsibility for any claims, instructions, methods or drug dosages contained in the abstracts. It is recommended that these are verified independently. The contents contained herein are correct at the time of printing and may be subject to change. Incorporating European Journal of Surgery and Swiss Surgery A Journal formed by the union of BJS, Acta Chirurgica Scandinavica, publisher of European Journal of Surgery, and the Swiss Society of Surgery, publisher of Swiss Surgery. The Journal is specially related to the Association of Surgeons of Great Britain and Ireland, Spanish Society of Surgical Research, Swedish Surgical Society and Swiss Society of Surgery. Editorial Team Editor-in-Chief Editors J. J. Earnshaw, Gloucester, UK J. Beynon, Swansea, UK C. H. C. Dejong, Maastricht, The Netherlands M. D. Evans, Swansea, UK R. J. Hinchliffe, Bristol, UK K. Søreide, Edinburgh, UK M. Sund, Umeå, Sweden B. P. L. Wijnhoven, Rotterdam, The Netherlands D. C. Winter, Dublin, Ireland Statistical Editors J. A. Cook, Oxford, UK J. Ranstam, Lund, Sweden Council Chairman Treasurer O. J. Garden, Edinburgh, UK M. -

A Multi-Disciplinary Model of Survivorship Care Following Definitive Chemoradiation for Anal Cancer Marissa B
Savoie et al. BMC Cancer (2019) 19:906 https://doi.org/10.1186/s12885-019-6053-y REVIEW Open Access A multi-disciplinary model of survivorship care following definitive chemoradiation for anal cancer Marissa B. Savoie1, Angela Laffan2, Cristina Brickman3, Bevin Daniels4, Anna Levin2,5, Tami Rowen6, James Smith7, Erin L. Van Blarigan2,7,8, Thomas A. Hope2,9, J. Michael Berry-Lawhorn10, Mekhail Anwar2,11 and Katherine Van Loon2,10* Abstract Following definitive chemoradiation for anal squamous cell carcinoma (ASCC), patients face a variety of chronic issues including: bowel dysfunction, accelerated bone loss, sexual dysfunction, and psychosocial distress. The increasing incidence of this disease, high cure rates, and significant long-term sequelae warrant increased focus on optimal survivorship care following definitive chemoradiation. In order to establish our survivorship care model for ASCC patients, a multi-disciplinary team of experts performed a comprehensive literature review and summarized best practices for the multi-disciplinary management of this unique patient population. We reviewed principle domains of our survivorship approach: (1) management of chronic toxicities; (2) sexual health; (3) HIV management in affected patients; (4) psychosocial wellbeing; and (5) surveillance for disease recurrence and survivorship care delivery. We provide recommendations for the optimization of survivorship care for ASCC patients can through a multi-disciplinary approach that supports physical and psychological wellness. Keywords: Anal cancer, Survivorship, Toxicity, Surveillance Background in women is comparable to that of cervical HPV in Anal squamous cell carcinoma (ASCC) is a rare cancer, women [7], and HPV transmission can occur during vagi- with only 8580 cases diagnosed in the United States annu- nal intercourse due to contamination of the entire perineal ally. -

Antiretroviral Therapy As a Factor Protective Against Anal Dysplasia in HIV-Infected Males Who Have Sex with Males C
721 Antiretroviral therapy as a factor protective against anal dysplasia in HIV-infected males who have sex with males C. Hidalgo Tenorio (a), C Gil Anguita (a), J. Ramírez Taboada(a), JG Martinez Cara (b), J. Esquivias (c), P Palma (d), M. Rivero (a) R Javier Martinez (a), MA Lopez Ruz (a), J. Pasquau Liaño (a). (a) Infectious Disease Unit; (b) Gastroenterology Service; (c) Pathology Service (d), General Surgery Service University Hospital Virgen de las Nieves, Granada Spain. Background: Studies show higher incidences of anal carcinoma and faster Material and Methods: A cross-sectional study of an HIV-positive MSM cohort. progression in HIV-infected MSM patients. Chronic infection with oncogenic HPV Epidemiological (condylomas, sexual habits, use of condom, smoking, alcohol consumption, is associated with the development of anal dysplasia. Antiretroviral therapy other infections, etc), clinical (months since HIV diagnosis, HIV stage according to CDC (ARV) has been shown to decrease the incidence of cervical carcinoma in criteria, ARV, virological failure), and analytical (Cd4 lymphocyte, Viral Load (VL), Cd4 nadir) women living with HIV; however, so far in the majority of publications ARV has data were collected. Two anal mucosa samples were taken and sent for HR-HPV PCR not proven to have a beneficial effect on the appearance of anal dysplastic testing and cytology. The genotypes 16, 18, 26, 31, 33, 35, 39, 45, 51–53, 56, 58, 59, 66, lesions, except in the Swiss cohort.Antiretroviral therapy (ARV) has been shown 68, 73 and 82 were considered high risk (HR-HPV). Genotypes 6, 11, 34, 40, 42–44, 54, 55, to decrease the incidence of cervical carcinoma in women with HIV.