Demonstrating the Importance of Membrane Repair in Response to Disease and Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functions of Vertebrate Ferlins
cells Review Functions of Vertebrate Ferlins Anna V. Bulankina 1 and Sven Thoms 2,* 1 Department of Internal Medicine 1, Goethe University Hospital Frankfurt, 60590 Frankfurt, Germany; [email protected] 2 Department of Child and Adolescent Health, University Medical Center Göttingen, 37075 Göttingen, Germany * Correspondence: [email protected] Received: 27 January 2020; Accepted: 20 February 2020; Published: 25 February 2020 Abstract: Ferlins are multiple-C2-domain proteins involved in Ca2+-triggered membrane dynamics within the secretory, endocytic and lysosomal pathways. In bony vertebrates there are six ferlin genes encoding, in humans, dysferlin, otoferlin, myoferlin, Fer1L5 and 6 and the long noncoding RNA Fer1L4. Mutations in DYSF (dysferlin) can cause a range of muscle diseases with various clinical manifestations collectively known as dysferlinopathies, including limb-girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy. A mutation in MYOF (myoferlin) was linked to a muscular dystrophy accompanied by cardiomyopathy. Mutations in OTOF (otoferlin) can be the cause of nonsyndromic deafness DFNB9. Dysregulated expression of any human ferlin may be associated with development of cancer. This review provides a detailed description of functions of the vertebrate ferlins with a focus on muscle ferlins and discusses the mechanisms leading to disease development. Keywords: dysferlin; myoferlin; otoferlin; C2 domain; calcium-sensor; muscular dystrophy; dysferlinopathy; limb girdle muscular dystrophy type 2B (LGMD2B), membrane repair; T-tubule system; DFNB9 1. Introduction Ferlins belong to the superfamily of proteins with multiple C2 domains (MC2D) that share common functions in tethering membrane-bound organelles or recruiting proteins to cellular membranes. Ferlins are described as calcium ions (Ca2+)-sensors for vesicular trafficking capable of sculpturing membranes [1–3]. -

Caveolin-1 Is Down-Regulated in Alveolar Rhabdomyosarcomas and Negatively Regulates Tumor Growth
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 5, No. 20 Caveolin-1 is down-regulated in alveolar rhabdomyosarcomas and negatively regulates tumor growth Juan Huertas-Martínez1, Santiago Rello-Varona1, David Herrero-Martín1, Ignasi Barrau1, Silvia García-Monclús1, Miguel Sáinz-Jaspeado1, Laura Lagares-Tena1, Yaiza Núñez-Álvarez5, Silvia Mateo-Lozano2, Jaume Mora2, Josep Roma3, Nuria Toran3, Sebastian Moran4, Roser López-Alemany1, Soledad Gallego3, Manel Esteller4, Miguel A. Peinado5, Xavier García del Muro1 and Oscar M. Tirado1 1 Sarcoma research group, Molecular Oncology Lab, Bellvitge Biomedical Research Institute (IDIBELL), L’Hospitalet de Llobregat, Barcelona, Spain 2 Developmental Tumor Biology Laboratory, Hospital Sant Joan de Deu, Barcelona, Spain 3 Biomedical Research Unit, Hospital Universitari Vall d’Hebron, Barcelona, Spain 4 Cancer Epigenetics and Biology Programme (PEBC), Bellvitge Biomedical Research Institute (IDIBELL), L’ Hospitalet de Llobregat, Barcelona, Spain 5 Institut de Medicina Predictiva i Personalitzada del Càncer, Badalona, Barcelona, Spain Correspondence to: Oscar M. Tirado, email: [email protected] Keywords: alveolar rhabdomyosarcoma, Caveolin-1, muscular differentiation, 5-AZA-2’-deoxycytidine, epigenetics, cell death Received: June 26, 2014 Accepted: August 26, 2014 Published: August 27, 2014 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood and adolescence. Despite advances in therapy, patients with histological variant of rhabdomyosarcoma known as alveolar rhabdomyosarcoma (ARMS) have a 5-year survival of less than 30%. Caveolin-1 (CAV1), encoding the structural component of cellular caveolae, is a suggested tumor suppressor gene involved in cell signaling. -

Novel Missense Mutation in the Caveolin-3 Gene in a Belgian Family
1349 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2003.028217 on 16 August 2004. Downloaded from SHORT REPORT Novel missense mutation in the caveolin-3 gene in a Belgian family with rippling muscle disease P Y K Van den Bergh, J M Ge´rard, J A Elosegi, M U Manto, C Kubisch, B G H Schoser ............................................................................................................................... J Neurol Neurosurg Psychiatry 2004;75:1349–1351. doi: 10.1136/jnnp.2003.028217 too small’’). The patient was diagnosed as having fibromyal- Rippling muscle disease (RMD) is a rare muscle disorder gia, which led to secondary depression and loss of her job. characterised by muscle stiffness, exercise induced myalgia, Muscle weakness and pigmenturia were absent. The medical and cramp-like sensations. It is genetically heterogeneous history was remarkable for mild hypothyroidism, peptic and can be acquired, but most cases show autosomal oesophagitis, and tobacco related asthmatiform bronchitis dominant inheritance due to mutations in the caveolin-3 with emphysema. At age 38, she had a neurological work-up. (CAV3) gene. We report a novel heterozygous missense The cranial nerves, muscle bulk, muscle strength, and deep mutation in CAV3 in a Belgian family with autosomal tendon reflexes were normal, and plantar responses were dominant RMD. flexor. Tapping with the reflex hammer or with the finger on A 40 year old woman complained of fatigue, exercise the muscles provoked an immediate, short lasting, forceful induced muscle pain, and muscle cramps since the age of 35. contraction and/or painful local mounding lasting for several Neurological examination revealed percussion induced seconds. PIRCs were most pronounced in the sternocleido- rapid muscle contractions (PIRCs) and localised muscle mastoid, deltoid, biceps, brachioradialis, finger and wrist mounding on percussion; muscle rippling was not observed. -

The Popeye Domain Containing Genes and Their Function in Striated Muscle
Journal of Cardiovascular Development and Disease Review The Popeye Domain Containing Genes and Their Function in Striated Muscle Roland F. R. Schindler 1, Chiara Scotton 2, Vanessa French 1, Alessandra Ferlini 2 and Thomas Brand 1,* 1 Developmental Dynamics, Harefield Heart Science Centre, National Heart and Lung Institute, Imperial College London, Hill End Road, Harefield UB9 6JH, UK; [email protected] (R.F.R.S.); [email protected] (V.F.) 2 Department of Medical Sciences, Medical Genetics Unit, University of Ferrara, Ferrara 44121, Italy; [email protected] (C.S.); fl[email protected] (A.F.) * Correspondence: [email protected]; Tel.: +44-189-582-8900 Academic Editors: Robert E. Poelmann and Monique R.M. Jongbloed Received: 26 April 2016; Accepted: 13 June 2016; Published: 15 June 2016 Abstract: The Popeye domain containing (POPDC) genes encode a novel class of cAMP effector proteins, which are abundantly expressed in heart and skeletal muscle. Here, we will review their role in striated muscle as deduced from work in cell and animal models and the recent analysis of patients carrying a missense mutation in POPDC1. Evidence suggests that POPDC proteins control membrane trafficking of interacting proteins. Furthermore, we will discuss the current catalogue of established protein-protein interactions. In recent years, the number of POPDC-interacting proteins has been rising and currently includes ion channels (TREK-1), sarcolemma-associated proteins serving functions in mechanical stability (dystrophin), compartmentalization (caveolin 3), scaffolding (ZO-1), trafficking (NDRG4, VAMP2/3) and repair (dysferlin) or acting as a guanine nucleotide exchange factor for Rho-family GTPases (GEFT). -
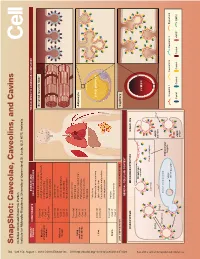
Snapshot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G
704 Cell SnapShot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G. Parton 154 Institute for Molecular Bioscience, University of Queensland, St. Lucia, QLD 4072, Australia , August 1,2013©2013 Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.07.009 ORGAN/ COMPONENTS DISEASE AND TISSUE-SPECIFIC CAVEOLAR COMPLEXES TISSUE CELLULAR PROCESS Rippling muscle disease Caveolin3 Striated muscle fiber Limb girdle muscular dystrophy Muscle Cavin1 skeletal and Cardiomyopathy Cavin4 cardiac Mechanoprotection Cav1 (heart) T-tubule formation Caveolin1 Lipodystrophy Caveolin2 Lipotoxicity Adipose Cavin1 Fatty acid regulation tissue Cavin2 Insulin signaling Cavin3 Mechanoprotection Atherosclerosis Caveolin1 Inammation Caveolin2 Lung Pulmonary hypertension and other Cavin1 Adipocyte Pulmonary brosis endothelia Cavin2 Mechanosensation Cavin3 Signaling Fatty liver Hepatocellular carcinoma Caveolin1 LIPID DROPLET Liver Lipid metabolism Caveolin2 Carbohydrate metabolism Liver regeneration Caveolin1 Autism Brain Cavin1 Schizophrenia Cavin3 LOW EXPRESSION HIGH EXPRESSION Capillary GENERAL CELLULAR CONTEXT ENDOCYTOSIS MECHANOPROTECTION SIGNALING See online version for legend and references. and legend for version online See Membrane stretch LUMEN Cavin complex Caveola EHD2 eNOS Ca2+ inactive Rab5 Dynamin2 EARLY ENDOSOME Intracellular targets MVB/ Caveolin 1 Caveolin 2 Caveolin 3 Dynamin2 late endosome eNOS Cavin1 Cavin2 Cavin3 Cavin4 eNOS EHD2 active SnapShot: Caveolae, Caveolins, and Cavins Nicholas Ariotti and Robert G. Parton Institute for Molecular Bioscience, University of Queensland, St. Lucia, QLD 4072, Australia Caveolae, submicroscopic bulb-shaped plasma membrane pits, are an abundant feature of many mammalian cells (Parton and del Pozo, 2013). Caveolae and the major proteins of caveolae, caveolins (Rothberg et al., 1992), and cavins (Hill et al., 2008), are linked to a number of human diseases such as muscular dystrophy, cardiomyopathy, and lipodys- trophy (see Glossary) (Bruno et al., 1993; Fernández et al., 2006; Hayashi et al., 2009). -

Development of an Infectious Clone System to Study the Life Cycle of Hazara Virus
Development of an Infectious Clone System to Study the Life Cycle of Hazara Virus Jack Fuller Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds Faculty of Biological Sciences July 2020 i The candidate confirms that the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. © 2020 The University of Leeds and Jack Fuller ii Acknowledgements I would like to thank my supervisors, John, Jamel and Roger for their endless support and fresh ideas throughout my PhD. A special thanks go to John for always providing enthusiasm and encouragement, especially during the tougher times of the project! I would also like to thank everyone in 8.61, not only for making my time at Leeds enjoyable, but for helping me develop as a scientist through useful critique and discussion. A special thanks goes to Francis Hopkins, for welcoming me into the Barr group and providing a constant supply of humour and to Ellie Todd, for listening to my endless whines and gripes, and for providing welcome distractions in the form of her PowerPoint artwork. Outside of the lab, my mum deserves a special mention for always being the voice of reason and support at the end of the phone, but also for pushing me to succeed throughout my entire education. I have no doubts I would not be in the position I am now without her! Finally, a huge thanks to my partner Hannah, who has been there to support me through all the highs and lows of my PhD, and has sacrificed many weekend trips to allow me to finish experiments and to tend to my viruses! iii Abstract Crimean-Congo hemorrhagic fever orthonairovirus (CCHFV) is a negative sense single stranded RNA virus, capable of causing fatal hemorrhagic fever in humans. -

Caveolar Endocytosis of Simian Virus 40 Reveals a New Two-Step Vesicular- Transport Pathway to the ER
articles Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular- transport pathway to the ER Lucas Pelkmans*, Jürgen Kartenbeck† and Ari Helenius*‡ *Institute of Biochemistry, Swiss Federal Institute of Technology, Universitaetstrasse 16, CH-8092 Zürich, Switzerland †German Cancer Research Center (DKFZ) Heidelberg, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany ‡e-mail: [email protected] Simian virus 40 (SV40) is unusual among animal viruses in that it enters cells through caveolae, and the internalized virus accumulates in a smooth endoplasmic reticulum (ER) compartment. Using video-enhanced, dual-colour, live fluorescence microscopy, we show the uptake of individual virus particles in CV-1 cells. After associating with cave- olae, SV40 leaves the plasma membrane in small, caveolin-1-containing vesicles. It then enters larger, peripheral organelles with a non-acidic pH. Although rich in caveolin-1, these organelles do not contain markers for endo- somes, lysosomes, ER or Golgi, nor do they acquire ligands of clathrin-coated vesicle endocytosis. After several hours in these organelles, SV40 is sorted into tubular, caveolin-free membrane vesicles that move rapidly along microtubules, and is deposited in perinuclear, syntaxin 17-positive, smooth ER organelles. The microtubule-disrupt- ing agent nocodazole inhibits formation and transport of these tubular carriers, and blocks viral infection. Our results demonstrate the existence of a two-step transport pathway from plasma-membrane caveolae, through an intermediate organelle (termed the caveosome), to the ER. This pathway bypasses endosomes and the Golgi com- plex, and is part of the productive infectious route used by SV40. any animal viruses take advantage of receptor-mediated mutants of caveolin-3 localize to intracellular vesicles that are dis- endocytosis to enter their host cells. -
Modulation of the Caveolin-3 Localization to Caveolae and STAT3 to Mitochondria by Catecholamine-Induced Cardiac Hypertrophy in H9c2 Cardiomyoblasts
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 41, No. 4, 226-235, April 2009 Modulation of the caveolin-3 localization to caveolae and STAT3 to mitochondria by catecholamine-induced cardiac hypertrophy in H9c2 cardiomyoblasts Kyuho Jeong*, Hayeong Kwon*, amine-induced cardiac hypertrophy. Chanhee Min and Yunbae Pak1 Keywords: cardiomegaly; caveolae; caveolin-3; cell Department of Biochemistry nucleus; heart; isoproterenol; mitochondria; phenyl- Division of Applied Life Science (BK21), PMBBRC ephrine; STAT3 transcription factor Environmental Biotechnology National Core Research Center Gyeongsang National University Jinju 660-701, Korea Introduction 1Corresponding author: Tel, 82-55-751-5961; Fax, 82-55-759-9363; E-mail, [email protected] Hypertension is major risk factors for cardiac da- mage, ischemia, myocardial infarction, and conge- *These authors contributed equally to this work. stive heart failure (Zampaglione et al., 1996). In DOI 10.3858/emm.2009.41.4.025 response to increased demands for cardiac work caused by various pathologic stresses, heart adapts Accepted 20 November 2008 through compensatory hypertrophy of myocytes. Abbreviations: Akt, protein kinase B; CsA, cyclosporin A; GPCR, Thus, cardiac hypertrophy is recognized in many G protein-coupled receptor; ISO, isoproterenol; PE, phenylephrine; cardiovascular diseases, such as hypertension, RTK, receptor tyrosine kinase; STAT3, signal transducers and acti- vascular disease, and myocardial infarction, and is vator of transcription 3 an independent risk factor for cardiac morbidity and mortality. Hypertrophic stimuli induce an in- crease in cell size in the absence of cell division through Ca2+ signaling and activation of PKC, Abstract MAPK and PKB/ Akt (Watanabe et al., 2001; Dorn and Force, 2005), and are accompanied by We investigated the effect of phenylephrine (PE)- and increased protein synthesis with reprogramming of isoproterenol (ISO)-induced cardiac hypertrophy on gene expression (Takeo et al., 2000). -

Reproductionresearch
REPRODUCTIONRESEARCH The association between CDC42 and caveolin-1 is involved in the regulation of capacitation and acrosome reaction of guinea pig and mouse sperm R Baltie´rrez-Hoyos, A L Roa-Espitia and E O Herna´ndez-Gonza´lez Departamento de Biologı´a Celular, Centro de Investigacio´n y Estudios Avanzados del Instituto Polite´cnico Nacional, Avenida Instituto Polite´cnico Nacional 2508, San Pedro Zacatenco, Me´xico DF 07360, Mexico Correspondence should be addressed to E O Herna´ndez-Gonza´lez; Email: [email protected] Abstract In the mammalian sperm, the acrosome reaction (AR) is considered to be a regulated secretion that is an essential requirement for physiological fertilization. The AR is the all-or-nothing secretion system that allows for multiple membrane fusion events. It is a C Ca2 -regulated exocytosis reaction that has also been shown to be regulated by several signaling pathways. CDC42 has a central role in the regulated exocytosis through the activation of SNARE proteins and actin polymerization. Furthermore, the lipid raft protein caveolin- 1 (CAV1) functions as a scaffold and guanine nucleotide dissociation inhibitor protein for CDC42, which is inactivated when associated with CAV1. CDC42 and other RHO proteins have been shown to localize in the acrosome region of mammalian sperm; however, their relationship with the AR is unknown. Here, we present the first evidence that CDC42 and CAV1 could be involved in the regulation of capacitation and the AR. Our findings show that CDC42 is activated early during capacitation, reaching an activation maximum after 20 min of capacitation. Spontaneous and progesterone-induced ARs were inhibited when sperm were capacitated in presence of secramine A, a specific CDC42 inhibitor. -

Immunolocalization of Caveolin-1 and Caveolin-3 in Monkey Skeletal, Cardiac and Uterine Smooth Muscles
CELL STRUCTURE AND FUNCTION 27: 375–382 (2002) © 2002 by Japan Society for Cell Biology Immunolocalization of Caveolin-1 and Caveolin-3 in Monkey Skeletal, Cardiac and Uterine Smooth Muscles ∗ Yasuko Hagiwara 1 , Yasushi Nishina2, Hiroshi Yorifuji2, and Tateki Kikuchi1 1Department of Animal Models for Human Disease, National Institute of Neuroscience, National Center of Neu- rology and Psychiatry, Kodaira, Tokyo 187-8502, Japan, and 2Department of Anatomy II, National Defence Medical College, Tokorozawa, Saitama 359-8513, Japan ABSTRACT. Caveolin, a 20–24 kDa integral membrane protein, is a principal component of caveolar domains. Caveolin-1 is expressed predominantly in endothelial cells, fibroblasts, and adipocytes, while the expression of caveolin-3 is confined to muscle cells. However, their localization in various muscles has not been well documented. Using double-immunofluorescence labeling and confocal laser microscopy, we examined the localization of caveolins-1 and 3 in adult monkey skeletal, cardiac and uterine smooth muscles and the co-immunolocalization of these caveolins with dystrophin, which is a product of the Duchenne muscular dystrophy gene. In the skeletal muscle tissue, caveolin-3 was localized along the sarcolemma except for the transverse tubules, and co- immunolocalized with dystrophin, whereas caveolin-1 was absent except in the blood vessels of the muscle tissue. In cardiac muscle cells, caveolins-1 and -3 and dystrophin were co-immunolocalized on the sarcolemma and transverse tubules. In uterine smooth muscle cells, caveolin-1, but not caveolin-3, was co-immunolocalized with dystrophin on the sarcolemma. Key words: caveolin/skeletal muscle/cardiac muscle/smooth muscle/dystrophin Caveolin is a major component of the caveolae which are caveolin-1 or -3. -

Biochemical and Pathological Changes Result from Mutated Caveolin-3 in Muscle José Andrés González Coraspe1, Joachim Weis1, Mary E
González Coraspe et al. Skeletal Muscle (2018) 8:28 https://doi.org/10.1186/s13395-018-0173-y RESEARCH Open Access Biochemical and pathological changes result from mutated Caveolin-3 in muscle José Andrés González Coraspe1, Joachim Weis1, Mary E. Anderson2, Ute Münchberg3, Kristina Lorenz3, Stephan Buchkremer1, Stephanie Carr4, René Peiman Zahedi3,5,6, Eva Brauers1, Hannah Michels4, Yoshihide Sunada7, Hanns Lochmüller4,8,9,10, Kevin P. Campbell2, Erik Freier3, Denisa Hathazi3† and Andreas Roos3*† Abstract Background: Caveolin-3 (CAV3) is a muscle-specific protein localized to the sarcolemma. It was suggested that CAV3 is involved in the connection between the extracellular matrix (ECM) and the cytoskeleton. Caveolinopathies often go along with increased CK levels indicative of sarcolemmal damage. So far, more than 40 dominant pathogenic mutations have been described leading to several phenotypes many of which are associated with a mis-localization of the mutant protein to the Golgi. Golgi retention and endoplasmic reticulum (ER) stress has been demonstrated for the CAV3 p.P104L mutation, but further downstream pathophysiological consequences remained elusive so far. Methods: We utilized a transgenic (p.P104L mutant) mouse model and performed proteomic profiling along with immunoprecipitation, immunofluorescence and immunoblot examinations (including examination of α- dystroglycan glycosylation), and morphological studies (electron and coherent anti-Stokes Raman scattering (CARS) microscopy) in a systematic investigation of molecular and subcellular events in p.P104L caveolinopathy. Results: Our electron and CARS microscopic as well as immunological studies revealed Golgi and ER proliferations along with a build-up of protein aggregates further characterized by immunoprecipitation and subsequent mass spectrometry. Molecular characterization these aggregates showed affection of mitochondrial and cytoskeletal proteins which accords with our ultra-structural findings. -

Protein Kinase C Β: a New Target Therapy to Prevent the Long-Term Atypical Antipsychotic-Induced Weight Gain
Neuropsychopharmacology (2017) 42, 1491–1501 © 2017 American College of Neuropsychopharmacology. All rights reserved 0893-133X/17 www.neuropsychopharmacology.org Protein Kinase C β: a New Target Therapy to Prevent the Long-Term Atypical Antipsychotic-Induced Weight Gain 1,6 2,6 1 1 1 3 Alessandro Rimessi , Chiara Pavan , Elli Ioannidi , Federica Nigro , Claudia Morganti , Alberto Brugnoli , 3 4 4 3 5 4,7 Francesco Longo , Chiara Gardin , Letizia Ferroni , Michele Morari , Vincenzo Vindigni , Barbara Zavan ,1,7 and Paolo Pinton* 1 Department of Morphology, Surgery and Experimental Medicine, Section of Pathology, Oncology and Experimental Biology, Laboratory for 2 Technologies of Advanced Therapies (LTTA), University of Ferrara, Ferrara, Italy; Unit of Psychiatry, Department of Neurosciences NPSRR, 3 University of Padua, Padua, Italy; Department of Medical Sciences, Section of Pharmacology, Neuroscience Center and National Institute of 4 5 Neuroscience, University of Ferrara, Ferrara, Italy; Department of Biomedical Sciences, University of Padua, Padua, Italy; Unit of Plastic Surgery, Department of Neurosciences NPSRR, University of Padua, Padua, Italy Antipsychotic drugs are currently used in clinical practice for a variety of mental disorders. Among them, clozapine is the most effective medication for treatment-resistant schizophrenia and is most helpful in controlling aggression and the suicidal behavior in schizophrenia and schizoaffective disorder. Although clozapine is associated with a low likelihood of extrapyramidal symptoms and other neurological side effects, it is well known for the weight gain and metabolic side effects, which expose the patient to a greater risk of cardiovascular disorders and premature death, as well as psychosocial issues, leading to non-adherence to therapy.