Undiagnosed Wilson's Disease and Fibromyalgia Masking
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

The Inherited Metabolic Disorders News
The Inherited Metabolic Disorders News Summer 2011 Volume 8 Issue 2 From the Editor I hope everyone is enjoying their summer thus far! Our 8th annual Metabolic Family Day and 7th annual Low In this Issue Protein Cooking Demonstration were once again a huge success. See the section ”What’s New” on page 9 for a full report of the events, as well as pictures. ♦ From the Editor…1 As always, your suggestions and stories are welcome. Please contact me by email: [email protected] or telephone 519-685-8453 if you wish to contribute to the ♦ From Dr. Chitra Prasad…1 newsletter. I hope everyone has a safe and happy summer! ♦ Personal Stories… 2 Janice Little ♦ Featured This Issue … 4 From Dr Chitra Prasad ♦ Suzanne’s Corner… 6 Dear Friends, ♦ What’s New… 8 Hope you all are having a wonderful summer. We had a great metabolic family workshop with around 198 registrants this year. This was truly phenomenal. Thanks to all the team members and ♦ Research & Presentations … 13 the families in the planning committee for doing such a fantastic job. I was really touched when one of our young metabolic patients told her parents that she would like to attend the ♦ How to Make a Donation… 14 metabolic family workshop! Please see some of the highlights of the metabolic workshop in the newsletter for those who could not make it. The speeches by our youth (Sadiq, Leanna and Laura) ♦ Contact Information … 15 were appreciated by everyone. Big thanks to Jill Tosswill (our previous social worker) who coordinated their talks. -

A Drug-Induced Cholestatic Pattern
Review articles Hepatotoxicity: A Drug-Induced Cholestatic Pattern Laura Morales M.,1 Natalia Vélez L.,1 Octavio Germán Muñoz M., MD.2 1 Medical Student in the Faculty of Medicine and Abstract the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia Although drug induced liver disease is a rare condition, it explains 40% to 50% of all cases of acute liver 2 Internist and Hepatologist at the Hospital Pablo failure. In 20% to 40% of the cases, the pattern is cholestatic and is caused by inhibition of the transporters Tobon Uribe and in the Gastrohepatology Group at that regulate bile synthesis. This reduction in activity is directly or indirectly mediated by drugs and their me- the Universidad de Antioquia in Medellín, Colombia tabolites and/or by genetic polymorphisms and other risk factors of the patient. Its manifestations range from ......................................... biochemical alterations in the absence of symptoms to acute liver failure and chronic liver damage. Received: 30-01-15 Although there is no absolute test or marker for diagnosis of this disease, scales and algorithms have Accepted: 26-01-16 been developed to assess the likelihood of cholestatic drug induced liver disease. Other types of evidence are not routinely used because of their complexity and cost. Diagnosis is primarily based on exclusion using circumstantial evidence. Cholestatic drug induced liver disease has better overall survival rates than other patters, but there are higher risks of developing chronic liver disease. In most cases, the patient’s condition improves when the drug responsible for the damage is removed. Hemodialysis and transplantation should be considered only for selected cases. -

Menkes Disease in 5 Siblings
Cu(e) the balancing act: Copper homeostasis explored in 5 siblings Poster with variable clinical course 595 Sonia A Varghese MD MPH MBA and Yael Shiloh-Malawsky MD Objective Methods Case Presentation Discussion v Present a unique variation of v Review literature describing v The graph below depicts the phenotypic spectrum seen in the 5 brothers v ATP7A mutations produce a clinical spectrum phenotype and course in siblings copper transport disorders v Mom is a known carrier of ATP7A mutation v Siblings 1, 2, and 5 follow a more classic with Menkes Disease (MD) v Apply findings to our case of five v There is limited information on the siblings who were not seen at UNC Menke’s course, while siblings 3 and 4 exhibit affected siblings v The 6th sibling is the youngest and is a healthy female infant (not included here) a phenotypic variation Sibling: v This variation is suggestive of a milder form of Background Treatment birth Presentation Exam Diagnostic testing Outcomes Copper Histidine Menkes such as occipital horn syndrome with v Mutations in ATP7A: copper deficiency order D: 16mos residual copper transport function (Menkes disease) O/AD: 1 Infancy/12mos N/A N/A No Brain v Siblings 3 and 4 had improvement with copper v Mutations in ATP7B: copper overload FTT, Seizures, DD hemorrhage supplementation, however declined when off (Wilson disease) O/AD: Infancy/NA D: 13mos supplementation -suggesting residual ATP7A v The amount of residual functioning 2 N/A N/A No FTT, FTT copper transport function copper transport influences disease Meningitis -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

Menkes Disease Submitted By: Alice Ho, Jean Mah, Robin Casey, Penney Gaul
Neuroimaging Highlight Editors: William Hu, Mark Hudon, Richard Farb “From Sheep to Babe” – Menkes Disease Submitted by: Alice Ho, Jean Mah, Robin Casey, Penney Gaul Can. J. Neurol. Sci. 2003; 30: 358-360 A four-month-old boy presented with a new onset focal On examination, his length (66 cm) and head circumference seizure lasting 18 minutes. During the seizure, his head and eyes (43 cm) were at the 50th percentile, and his weight (6.6 kg) was were deviated to the left, and he assumed a fencing posture to the at the 75th percentile. He had short bristly pale hair, and a left with pursing of his lips. He had been unwell for one week, cherubic appearance with full cheeks. His skin was velvety and with episodes of poor feeding, during which he would become lax. He was hypotonic with significant head lag and slip-through unresponsive, limp and stare for a few seconds. Developmentally on vertical suspension. His cranial nerves were intact. He moved he was delayed. He was not yet rolling, and he had only just all limbs equally well with normal strength. His deep tendon begun to lift his head in prone position. He could grasp but was reflexes were 3+ at patellar and brachioradialis, and 2+ not reaching or bringing his hands together at the midline. He elsewhere. Plantar responses were upgoing. was cooing but not laughing. His past medical history was Laboratory investigations showed increased lactate (7.7 significant for term delivery with fetal distress and meconium mmol/L) and alkaline phosphatase (505 U/L), with normal CBC, staining. -

Menkes Disease: Report of Two Cases
Iran J Pediatr Case Report Dec 2007; Vol 17 ( No 3), Pp:388-392 Menkes Disease: Report of Two Cases Mohammad Barzegar*1, MD; Afshin Fayyazie1, MD; Bobollah Gasemie2, MD; Mohammad ali Mohajel Shoja3, MD 1. Pediatric Neurologist, Department of Pediatrics, Tabriz University of Medical Sciences, IR Iran 2. Pathologist, Department of Pathology, Tabriz University of Medical Sciences, IR Iran 3. General Physician, Tabriz University of Medical Sciences, IR Iran Received: 14/05/07; Revised: 20/08/07; Accepted: 10/10/07 Abstract Introduction: Menkes disease is a rare X-linked recessive disorder of copper metabolism. It is characterized by progressive cerebral degeneration with psychomotor deterioration, hypothermia, seizures and characteristic facial appearance with hair abnormalities. Case Presentation: We report on two cases of classical Menkes disease with typical history, (progressive psychomotor deterioration and seizures}, clinical manifestations (cherubic appearance, with brittle, scattered and hypopigmented scalp hairs), and progression. Light microscopic examination of the hair demonstrated the pili torti pattern. The low serum copper content and ceruloplasmin confirmed the diagnosis. Conclusion: Menkes disease is an under-diagnosed entity, being familiar with its manifestation and maintaining high index of suspicion are necessary for early diagnosis. Key Words: Menkes disease, Copper metabolism, Epilepsy, Pili torti, Cerebral degeneration Introduction colorless. Examination under microscope reveals Menkes disease (MD), also referred to as kinky a variety of abnormalities, most often pili torti hair disease, trichopoliodystrophy, and steely hair (twisted hair), monilethrix (varying diameter of disease, is a rare X-linked recessive disorder of hair shafts) and trichorrhexis nodosa (fractures of copper metabolism.[1,2] It is characterized by the hair shaft at regular intervals)[4]. -
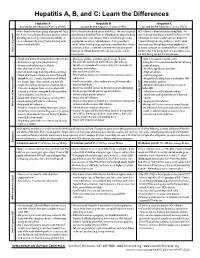
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Multipoint Linkage Analysis in Menkes Disease T
Am. J. Hum. Genet. 50:1012-1017, 1992 Multipoint Linkage Analysis in Menkes Disease T. T0nnesen,* A. Petterson,* T. A. KruseT A.-M. Gerdest and N. Horn* *John F. Kennedy Institute, Glostrup, Denmark; TInstitute of Human Genetics, University of Aarhus, Aarhus, Denmark; and *Department of Clinical Chemistry, Odense Sygehus, Odense, Denmark Summary Linkage analyses were performed in 11 families with X-linked Menkes disease. In each family more than one affected patient had been diagnosed. Forty informative meioses were tested using 11 polymorphic DNA markers. From two-point linkage analyses high lod scores are seen for DXS146 (pTAK-8; maximal lod score 3.16 at recombination fraction [0] = .0), for DXS1 (p-8; maximal lod score 3.44 at 0 = .0), for PGK1 (maximal lod score 2.48 at 0 = .0), and for DXS3 (p19-2; maximal lod score 2.90 at 0 = .0). This indicates linkage to the pericentromeric region. Multilocus linkage analyses of the same data revealed a peak for the location score between DXS146(pTAK-8) and DXYSlX(pDP34). The most likely location is between DXS159 (cpX289) and DXYS1X(pDP34). Odds for this location relative to the second-best-supported region, be- tween DXS146(pTAK-8) and DXS159 (cpX289), are better than 74:1. Visualization of individual recombi- nant X chromosomes in two of the Menkes families showed the Menkes locus to be situated between DXS159(cpX289) and DXS94(pXG-12). Combination of the present results with the reported absence of Menkes symptoms in male patients with deletions in Xq21 leads to the conclusion that the Menkes locus is proximal to DXSYlX(pDP34) and located in the region Xql2 to Xql3.3. -

Diagnosis and Management of Primary Biliary Cholangitis Ticle
REVIEW ArtICLE 1 see related editorial on page x Diagnosis and Management of Primary Biliary Cholangitis TICLE R Zobair M. Younossi, MD, MPH, FACG, AGAF, FAASLD1, David Bernstein, MD, FAASLD, FACG, AGAF, FACP2, Mitchell L. Shifman, MD3, Paul Kwo, MD4, W. Ray Kim, MD5, Kris V. Kowdley, MD6 and Ira M. Jacobson, MD7 Primary biliary cholangitis (PBC) is a chronic, cholestatic, autoimmune disease with a variable progressive course. PBC can cause debilitating symptoms including fatigue and pruritus and, if left untreated, is associated with a high risk of cirrhosis and related complications, liver failure, and death. Recent changes to the PBC landscape include a REVIEW A name change, updated guidelines for diagnosis and treatment as well as new treatment options that have recently become available. Practicing clinicians face many unanswered questions when managing PBC. To assist these healthcare providers in managing patients with PBC, the American College of Gastroenterology (ACG) Institute for Clinical Research & Education, in collaboration with the Chronic Liver Disease Foundation (CLDF), organized a panel of experts to evaluate and summarize the most current and relevant peer-reviewed literature regarding PBC. This, combined with the extensive experience and clinical expertise of this expert panel, led to the formation of this clinical guidance on the diagnosis and management of PBC. Am J Gastroenterol https://doi.org/10.1038/s41395-018-0390-3 INTRODUCTION addition, diagnosis and treatment guidelines are changing and a Primary biliary cholangitis (PBC) is a chronic, cholestatic, auto- number of guidelines have been updated [4, 5]. immune disease with a progressive course that may extend over Because of important changes in the PBC landscape, and a num- many decades. -

Guideline for the Evaluation of Cholestatic Jaundice
CLINICAL GUIDELINES Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition ÃRima Fawaz, yUlrich Baumann, zUdeme Ekong, §Bjo¨rn Fischler, jjNedim Hadzic, ôCara L. Mack, #Vale´rie A. McLin, ÃÃJean P. Molleston, yyEzequiel Neimark, zzVicky L. Ng, and §§Saul J. Karpen ABSTRACT Cholestatic jaundice in infancy affects approximately 1 in every 2500 term PREAMBLE infants and is infrequently recognized by primary providers in the setting of holestatic jaundice in infancy is an uncommon but poten- physiologic jaundice. Cholestatic jaundice is always pathologic and indicates tially serious problem that indicates hepatobiliary dysfunc- hepatobiliary dysfunction. Early detection by the primary care physician and tion.C Early detection of cholestatic jaundice by the primary care timely referrals to the pediatric gastroenterologist/hepatologist are important physician and timely, accurate diagnosis by the pediatric gastro- contributors to optimal treatment and prognosis. The most common causes of enterologist are important for successful treatment and an optimal cholestatic jaundice in the first months of life are biliary atresia (25%–40%) prognosis. The Cholestasis Guideline Committee consisted of 11 followed by an expanding list of monogenic disorders (25%), along with many members of 2 professional societies: the North American Society unknown or multifactorial (eg, parenteral nutrition-related) causes, each of for Gastroenterology, Hepatology and Nutrition, and the European which may have time-sensitive and distinct treatment plans. Thus, these Society for Gastroenterology, Hepatology and Nutrition. This guidelines can have an essential role for the evaluation of neonatal cholestasis committee has responded to a need in pediatrics and developed to optimize care. -

Clinical Expression of Menkes Disease in Females with Normal Karyotype
Møller et al. Orphanet Journal of Rare Diseases 2012, 7:6 http://www.ojrd.com/content/7/1/6 RESEARCH Open Access Clinical expression of Menkes disease in females with normal karyotype Lisbeth Birk Møller1*, Malgorzata Lenartowicz2, Marie-Therese Zabot3, Arnaud Josiane4, Lydie Burglen5, Chris Bennett6, Daniel Riconda7, Richard Fisher8, Sandra Janssens9, Shehla Mohammed10, Margreet Ausems11, Zeynep Tümer1, Nina Horn1 and Thomas G Jensen12 Abstract Background: Menkes Disease (MD) is a rare X-linked recessive fatal neurodegenerative disorder caused by mutations in the ATP7A gene, and most patients are males. Female carriers are mosaics of wild-type and mutant cells due to the random X inactivation, and they are rarely affected. In the largest cohort of MD patients reported so far which consists of 517 families we identified 9 neurologically affected carriers with normal karyotypes. Methods: We investigated at-risk females for mutations in the ATP7A gene by sequencing or by multiplex ligation- dependent probe amplification (MLPA). We analyzed the X-inactivation pattern in affected female carriers, unaffected female carriers and non-carrier females as controls, using the human androgen-receptor gene methylation assay (HUMAR). Results: The clinical symptoms of affected females are generally milder than those of affected boys with the same mutations. While a skewed inactivation of the X-chromosome which harbours the mutation was observed in 94% of 49 investigated unaffected carriers, a more varied pattern was observed in the affected carriers. Of 9 investigated affected females, preferential silencing of the normal X-chromosome was observed in 4, preferential X-inactivation of the mutant X chromosome in 2, an even X-inactivation pattern in 1, and an inconclusive pattern in 2.