Characterizing Rumen Microbiota and Cazyme Profile of Indian
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fossil Bovidae from the Malay Archipelago and the Punjab
FOSSIL BOVIDAE FROM THE MALAY ARCHIPELAGO AND THE PUNJAB by Dr. D. A. HOOIJER (Rijksmuseum van Natuurlijke Historie, Leiden) with pls. I-IX CONTENTS Introduction 1 Order Artiodactyla Owen 8 Family Bovidae Gray 8 Subfamily Bovinae Gill 8 Duboisia santeng (Dubois) 8 Epileptobos groeneveldtii (Dubois) 19 Hemibos triquetricornis Rütimeyer 60 Hemibos acuticornis (Falconer et Cautley) 61 Bubalus palaeokerabau Dubois 62 Bubalus bubalis (L.) subsp 77 Bibos palaesondaicus Dubois 78 Bibos javanicus (d'Alton) subsp 98 Subfamily Caprinae Gill 99 Capricornis sumatraensis (Bechstein) subsp 99 Literature cited 106 Explanation of the plates 11o INTRODUCTION The Bovidae make up a very large portion of the Dubois collection of fossil vertebrates from Java, second only to the Proboscidea in bulk. Before Dubois began his explorations in Java in 1890 we knew very little about the fossil bovids of that island. Martin (1887, p. 61, pl. VII fig. 2) described a horn core as Bison sivalensis Falconer (?); Bison sivalensis Martin has al• ready been placed in the synonymy of Bibos palaesondaicus Dubois by Von Koenigswald (1933, p. 93), which is evidently correct. Pilgrim (in Bron- gersma, 1936, p. 246) considered the horn core in question to belong to a Bibos species closely related to the banteng. Two further horn cores from Java described by Martin (1887, p. 63, pl. VI fig. 4; 1888, p. 114, pl. XII fig. 4) are not sufficiently well preserved to allow of a specific determination, although they probably belong to Bibos palaesondaicus Dubois as well. In a preliminary faunal list Dubois (1891) mentions four bovid species as occurring in the Pleistocene of Java, viz., two living species (the banteng and the water buffalo) and two extinct forms, Anoa spec. -

Bison Literature Review Biology
Bison Literature Review Ben Baldwin and Kody Menghini The purpose of this document is to compare the biology, ecology and basic behavior of cattle and bison for a management context. The literature related to bison is extensive and broad in scope covering the full continuum of domestication. The information incorporated in this review is focused on bison in more or less “wild” or free-ranging situations i.e.., not bison in close confinement or commercial production. While the scientific literature provides a solid basis for much of the basic biology and ecology, there is a wealth of information related to management implications and guidelines that is not captured. Much of the current information related to bison management, behavior (especially social organization) and practical knowledge is available through local experts, current research that has yet to be published, or popular literature. These sources, while harder to find and usually more localized in scope, provide crucial information pertaining to bison management. Biology Diet Composition Bison evolutional history provides the basis for many of the differences between bison and cattle. Bison due to their evolution in North America ecosystems are better adapted than introduced cattle, especially in grass dominated systems such as prairies. Many of these areas historically had relatively low quality forage. Bison are capable of more efficient digestion of low-quality forage then cattle (Peden et al. 1973; Plumb and Dodd 1993). Peden et al. (1973) also found that bison could consume greater quantities of low protein and poor quality forage then cattle. Bison and cattle have significant dietary overlap, but there are slight differences as well. -

Last Interglacial (MIS 5) Ungulate Assemblage from the Central Iberian Peninsula: the Camino Cave (Pinilla Del Valle, Madrid, Spain)
Palaeogeography, Palaeoclimatology, Palaeoecology 374 (2013) 327–337 Contents lists available at SciVerse ScienceDirect Palaeogeography, Palaeoclimatology, Palaeoecology journal homepage: www.elsevier.com/locate/palaeo Last Interglacial (MIS 5) ungulate assemblage from the Central Iberian Peninsula: The Camino Cave (Pinilla del Valle, Madrid, Spain) Diego J. Álvarez-Lao a,⁎, Juan L. Arsuaga b,c, Enrique Baquedano d, Alfredo Pérez-González e a Área de Paleontología, Departamento de Geología, Universidad de Oviedo, C/Jesús Arias de Velasco, s/n, 33005 Oviedo, Spain b Centro Mixto UCM-ISCIII de Evolución y Comportamiento Humanos, C/Sinesio Delgado, 4, 28029 Madrid, Spain c Departamento de Paleontología, Facultad de Ciencias Geológicas, Universidad Complutense de Madrid, Ciudad Universitaria, 28040 Madrid, Spain d Museo Arqueológico Regional de la Comunidad de Madrid, Plaza de las Bernardas, s/n, 28801-Alcalá de Henares, Madrid, Spain e Centro Nacional de Investigación sobre la Evolución Humana (CENIEH), Paseo Sierra de Atapuerca, s/n, 09002 Burgos, Spain article info abstract Article history: The fossil assemblage from the Camino Cave, corresponding to the late MIS 5, constitutes a key record to un- Received 2 November 2012 derstand the faunal composition of Central Iberia during the last Interglacial. Moreover, the largest Iberian Received in revised form 21 January 2013 fallow deer fossil population was recovered here. Other ungulate species present at this assemblage include Accepted 31 January 2013 red deer, roe deer, aurochs, chamois, wild boar, horse and steppe rhinoceros; carnivores and Neanderthals Available online 13 February 2013 are also present. The origin of the accumulation has been interpreted as a hyena den. Abundant fallow deer skeletal elements allowed to statistically compare the Camino Cave fossils with other Keywords: Early Late Pleistocene Pleistocene and Holocene European populations. -

Conservation Status of Asiatic Wild Buffalo (Bubalus Arnee) in Chhattisgarh
Conservation status of Asiatic Wild Buffalo (Bubalus arnee) in Chhattisgarh revealed through genetic study A Technical Report Prepared by Laboratory for the Conservation of Wildlife Trust of India Endangered Species(LACONES) F – 13, Sector 08 CSIR-CCMB Annex I, HYDERABAD – 500048 NCR, Noida - 201301 Disclaimer: This publication is meant for authorized use by laboratories and persons involved in research on conservation of Wild buffalos. LaCONES shall not be liable for any direct, consequential or incidental damages arising out of the protocols described in this book. Reference to any specific product (commercial or non-commercial), processes or services by brand or trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation or favor by LaCONES. The information and statements contained in this document shall not be used for the purpose of advertising or to imply the endorsement or recommendation of LaCONES. Citation: Mishra R.P. and A. Gaur. 2019. Conservation status of Asiatic Wild Buffalo (Bubalus arnee) in Chhattisgarh revealed through genetic study. Technical Report of WTI and CSIR-CCMB, 17p ACKNOWLEDGEMENTS We are thankful to the Forest Department, Govt. of Chhattisgarh for giving permission to carry out the conservation and research activities on Wild buffalo in various protected areas in Chhattisgarh. We are grateful to Shri Ram Prakash, PCCF (Retd.); Shri R.N. Mishra, PCCF (Retd.); Dr. R.K. Singh, PCCF (Retd.), Shri Atul Kumar Shukla, Principal Chief Conservator of Forests & Chief Wildlife Warden and Dr. S.K. Singh, Additional Principal Chief Conservator of Forests (WL), Dr. Rakesh Mishra, Director CSIR-CCMB, Dr. Rahul Kaul, Executive Director, WTI, Dr. -

A Bubaline-Derived Satellite DNA Probe Uncovers Generic Affinities of Gaur with Other Bovids
A bubaline-derived satellite DNA probe uncovers generic affinities of gaur with other bovids PRITHA RAY, SUPRIYA GANGADHARAN, MUNMUN CHATrOPADHYAY, ANU BASHAMBOO, SUN1TA BHATNAGAR, PRADEEP KUMAR MALIK* and SHER ALI t Molecular Genetics Laboratory, National Institute of Immunology, Aruna Asaf Ali Marg, New Delhi 110 067, India *Wildlife Institute of lndia, Chandrabani, Dehra Dun 248 001, India tCorresponding author (Fax, 91-11-6162125; Email, [email protected]). DNA typing using genome derived cloned probes may be conducted for ascertaining genetic affinities of closely related species. We analysed gaur Bos gaurus, cattle Bos indicus, buffalo Bubalus bubalis, sheep Ovis aries and goat Capra hircus DNA using buffalo derived cloned probe pDS5 carrying an array of BamHI satellite fraction of 1378 base residues to uncover its genomic organization. Zoo-blot analysis showed that pDS5 does not cross hybridize with non-bovid animals and surprisingly with female gaur genomic DNA. The presence of pDS5 sequences in the gaur males suggests their possible location on the Y chromosome. Genotyping of pDS5 with BamHI enzyme detected mostly monomorphic bands in the bubaline samples and polymorphic ones in cattle and gaur giving rise to clad specific pattern. Similar typing with RsaI enzyme also revealed clad specific band pattern detecting more number of bands in buffalo and fewer in sheep, goat and gaur samples. Copy number variation was found to be prominent in cattle and gaur with RsaI typing. Our data based on matched band profiles (MBP) suggest that gaur is genetically closer to cattle than buffalo contradicting the age-old notion held by some that gaur is a wild buffalo. -

Vega Etal Procroyalsocb Synchronous Diversification
Canterbury Christ Church University’s repository of research outputs http://create.canterbury.ac.uk Please cite this publication as follows: Frantz, Laurent A. F., Rudzinski, A., Mansyursyah Surya Nugraha, A., Evin, A., Burton, J., Hulme-Beaman, A., Linderholm, A., Barnett, R., Vega, R., Irving-Pease, E., Haile, J., Allen, R., Leus, K., Shephard, J., Hillyer, M., Gillemot, S., van den Hurk, J., Ogle, S., Atofanei, C., Thomas, M., Johansson, F., Haris Mustari, A., Williams, J., Mohamad, K., Siska Damayanti, C., Djuwita Wiryadi, I., Obbles, D., Mona, S., Day, H., Yasin, M., Meker, S., McGuire, J., Evans, B., von Rintelen, T., Hoult, S., Searle, J., Kitchener, A., Macdonald, A., Shaw, D., Hall, R., Galbusera, P. and Larson, G. (2018) Synchronous diversification of Sulawesi’s iconic artiodactyls driven by recent geological events. Proceedings of the Royal Society B: Biological Sciences. Link to official URL (if available): http://dx.doi.org/10.1098/rspb.2017.2566. This version is made available in accordance with publishers’ policies. All material made available by CReaTE is protected by intellectual property law, including copyright law. Any use made of the contents should comply with the relevant law. Contact: [email protected] Synchronous diversification of Sulawesi’s iconic artiodactyls driven by recent geological events Authors Laurent A. F. Frantz1,2,a,*, Anna Rudzinski3,*, Abang Mansyursyah Surya Nugraha4,c,*, , Allowen Evin5,6*, James Burton7,8*, Ardern Hulme-Beaman2,6, Anna Linderholm2,9, Ross Barnett2,10, Rodrigo Vega11 Evan K. Irving-Pease2, James Haile2,10, Richard Allen2, Kristin Leus12,13, Jill Shephard14,15, Mia Hillyer14,16, Sarah Gillemot14, Jeroen van den Hurk14, Sharron Ogle17, Cristina Atofanei11, Mark G. -
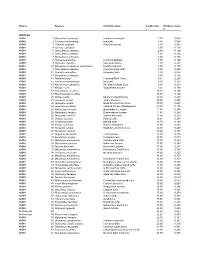
PDF File Containing Table of Lengths and Thicknesses of Turtle Shells And
Source Species Common name length (cm) thickness (cm) L t TURTLES AMNH 1 Sternotherus odoratus common musk turtle 2.30 0.089 AMNH 2 Clemmys muhlenbergi bug turtle 3.80 0.069 AMNH 3 Chersina angulata Angulate tortoise 3.90 0.050 AMNH 4 Testudo carbonera 6.97 0.130 AMNH 5 Sternotherus oderatus 6.99 0.160 AMNH 6 Sternotherus oderatus 7.00 0.165 AMNH 7 Sternotherus oderatus 7.00 0.165 AMNH 8 Homopus areolatus Common padloper 7.95 0.100 AMNH 9 Homopus signatus Speckled tortoise 7.98 0.231 AMNH 10 Kinosternon subrabum steinochneri Florida mud turtle 8.90 0.178 AMNH 11 Sternotherus oderatus Common musk turtle 8.98 0.290 AMNH 12 Chelydra serpentina Snapping turtle 8.98 0.076 AMNH 13 Sternotherus oderatus 9.00 0.168 AMNH 14 Hardella thurgi Crowned River Turtle 9.04 0.263 AMNH 15 Clemmys muhlenbergii Bog turtle 9.09 0.231 AMNH 16 Kinosternon subrubrum The Eastern Mud Turtle 9.10 0.253 AMNH 17 Kinixys crosa hinged-back tortoise 9.34 0.160 AMNH 18 Peamobates oculifers 10.17 0.140 AMNH 19 Peammobates oculifera 10.27 0.140 AMNH 20 Kinixys spekii Speke's hinged tortoise 10.30 0.201 AMNH 21 Terrapene ornata ornate box turtle 10.30 0.406 AMNH 22 Terrapene ornata North American box turtle 10.76 0.257 AMNH 23 Geochelone radiata radiated tortoise (Madagascar) 10.80 0.155 AMNH 24 Malaclemys terrapin diamondback terrapin 11.40 0.295 AMNH 25 Malaclemys terrapin Diamondback terrapin 11.58 0.264 AMNH 26 Terrapene carolina eastern box turtle 11.80 0.259 AMNH 27 Chrysemys picta Painted turtle 12.21 0.267 AMNH 28 Chrysemys picta painted turtle 12.70 0.168 AMNH 29 -

Patterns of Animal Utilization in the Holocene of the Philippines: a Comparison of Faunal Samples from Four Archaeological Sites
Patterns of Animal Utilization in the Holocene of the Philippines: A Comparison of Faunal Samples from Four Archaeological Sites KAREN M. MUDAR THE PHILIPPINE ARCHIPELAGO IS A SERIES OF TROPICAL OCEANIC ISLANDS located off the eastern edge of the Sunda Shelf While dramatically lower sea levels of 145-160 m during the middle and late Pleistocene uncovered the shelf and joined the Malay Peninsula and islands of Sumatra, Java, and Borneo into one land mass (Heaney 1985), this had little effect on accessibility to the oceanic Philippine Islands. Although relatively narrow water gaps, 12-25 km in width, existed between the archipelago and the Sunda Shelf at several times during the middle and late Pleistocene, the only major island that was connected to the mainland was Palawan,l which formed part of Sundaland during the middle Pleis tocene. Therefore nonarboreal mammalian fauna entering the archipelago either swam or were rafted to the oceanic islands. This isolation, combined with the relatively small size of the islands, has had a significant effect on the mammalian fauna, affecting both species diversity and morphology. The water barrier acted as a filter, allowing migration of small mam mals, particularly murid rodents, and discouraging migration of other taxa. Small insectivores, for example, have high metabolic requirements, which make it un likely that they would survive a lengthy rafting event. Other research on island biogeography (Heaney 1984) has shown that small isolated islands, in general, support a fauna depauperate in carnivores and large herbivores. In contrast to the 11 species of ungulates and 29 species of carnivores on Sumatra, the Philippines supports only three species of ungulates and two species of carnivores (Heaney 1984). -

Detomidine and Butorphanol for Standing Sedation in a Range of Zoo-Kept Ungulate Species
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Ghent University Academic Bibliography Journal of Zoo and Wildlife Medicine 48(3): 616–626, 2017 Copyright 2017 by American Association of Zoo Veterinarians DETOMIDINE AND BUTORPHANOL FOR STANDING SEDATION IN A RANGE OF ZOO-KEPT UNGULATE SPECIES Tim Bouts, D.V.M., M.Sc., Dip. E.C.Z.M., Joanne Dodds, V.N., Karla Berry, V.N., Abdi Arif, M.V.Sc., Polly Taylor, Vet. M. B., Ph. D., Dip. E.C.V.A.A., Andrew Routh, B. V. Sc., Cert. Zoo. Med., and Frank Gasthuys, D.V.M., Ph. D., Dip. E.C.V.A.A. Abstract: General anesthesia poses risks for larger zoo species, like cardiorespiratory depression, myopathy, and hyperthermia. In ruminants, ruminal bloat and regurgitation of rumen contents with potential aspiration pneumonia are added risks. Thus, the use of sedation to perform minor procedures is justified in zoo animals. A combination of detomidine and butorphanol has been routinely used in domestic animals. This drug combination, administered by remote intramuscular injection, can also be applied for standing sedation in a range of zoo animals, allowing a number of minor procedures. The combination was successfully administered in five species of nondomesticated equids (Przewalski horse [Equus ferus przewalskii; n ¼ 1], onager [Equus hemionus onager; n ¼ 4], kiang [Equus kiang; n ¼ 3], Grevy’s zebra [Equus grevyi; n ¼ 4], and Somali wild ass [Equus africanus somaliensis; n ¼ 7]), with a mean dose range of 0.10–0.17 mg/kg detomidine and 0.07–0.13 mg/kg butorphanol; the white (Ceratotherium simum simum; n ¼ 12) and greater one-horned rhinoceros (Rhinoceros unicornis; n ¼ 4), with a mean dose of 0.015 mg/kg of both detomidine and butorphanol; and Asiatic elephant bulls (Elephas maximus; n ¼ 2), with a mean dose of 0.018 mg/kg of both detomidine and butorphanol. -

European Bison
IUCN/Species Survival Commission Status Survey and Conservation Action Plan The Species Survival Commission (SSC) is one of six volunteer commissions of IUCN – The World Conservation Union, a union of sovereign states, government agencies and non- governmental organisations. IUCN has three basic conservation objectives: to secure the conservation of nature, and especially of biological diversity, as an essential foundation for the future; to ensure that where the Earth’s natural resources are used this is done in a wise, European Bison equitable and sustainable way; and to guide the development of human communities towards ways of life that are both of good quality and in enduring harmony with other components of the biosphere. A volunteer network comprised of some 8,000 scientists, field researchers, government officials Edited by Zdzis³aw Pucek and conservation leaders from nearly every country of the world, the SSC membership is an Compiled by Zdzis³aw Pucek, Irina P. Belousova, unmatched source of information about biological diversity and its conservation. As such, SSC Ma³gorzata Krasiñska, Zbigniew A. Krasiñski and Wanda Olech members provide technical and scientific counsel for conservation projects throughout the world and serve as resources to governments, international conventions and conservation organisations. IUCN/SSC Action Plans assess the conservation status of species and their habitats, and specifies conservation priorities. The series is one of the world’s most authoritative sources of species conservation information -

(Bubalus Bubalis) in NEPAL: RECOMMENDED MANAGEMENT ACTION in the FACE of UNCERTAINTY for a CRITICALLY ENDANGERED SPECIES
Contents TIGERPAPER A Translocation Proposal for Wild Buffalo in Nepal................... 1 Eucalyptus – Bane or Boon?................................................... 8 Status and Distribution of Wild Cattle in Cambodia.................... 9 Reptile Richness and Diversity In and Around Gir Forest........... 15 A Comparison of Identification Techniques for Predators on Artificial Nests................................................................... 20 Devastating Flood in Kaziranga National Park............................ 24 Bird Damage to Guava and Papaya........................................... 27 Death of an Elephant by Sunstroke in Orissa............................. 31 Msc in Forest and Nature Conservation for Tropical Areas......... 32 FOREST NEWS Report of an International Conference on Community Involvement in Fire Management............................................ 1 ASEAN Senior Officials Endorse Code of Practice for Forest Harvesting.................................................................. 4 Asian Model Forests Develop Criteria and Indicators Guidelines............................................................................. 4 East Asian Countries Pledge Action on Illegal Forest Activities.............................................................................. 6 South Pacific Ministers Consider Forestry Issues........................ 9 Tropical Ecosystems, Structure, Diversity and Human Welfare.. 10 Draft Webpage for International Weem Network......................... 10 New FAO Forestry Publications............................................... -

Pan-American Trypanosoma (Megatrypanum) Trinaperronei N. Sp
Garcia et al. Parasites Vectors (2020) 13:308 https://doi.org/10.1186/s13071-020-04169-0 Parasites & Vectors RESEARCH Open Access Pan-American Trypanosoma (Megatrypanum) trinaperronei n. sp. in the white-tailed deer Odocoileus virginianus Zimmermann and its deer ked Lipoptena mazamae Rondani, 1878: morphological, developmental and phylogeographical characterisation Herakles A. Garcia1,2* , Pilar A. Blanco2,3,4, Adriana C. Rodrigues1, Carla M. F. Rodrigues1,5, Carmen S. A. Takata1, Marta Campaner1, Erney P. Camargo1,5 and Marta M. G. Teixeira1,5* Abstract Background: The subgenus Megatrypanum Hoare, 1964 of Trypanosoma Gruby, 1843 comprises trypanosomes of cervids and bovids from around the world. Here, the white-tailed deer Odocoileus virginianus (Zimmermann) and its ectoparasite, the deer ked Lipoptena mazamae Rondani, 1878 (hippoboscid fy), were surveyed for trypanosomes in Venezuela. Results: Haemoculturing unveiled 20% infected WTD, while 47% (7/15) of blood samples and 38% (11/29) of ked guts tested positive for the Megatrypanum-specifc TthCATL-PCR. CATL and SSU rRNA sequences uncovered a single species of trypanosome. Phylogeny based on SSU rRNA and gGAPDH sequences tightly cluster WTD trypanosomes from Venezuela and the USA, which were strongly supported as geographical variants of the herein described Trypa- nosoma (Megatrypanum) trinaperronei n. sp. In our analyses, the new species was closest to Trypanosoma sp. D30 from fallow deer (Germany), both nested into TthII alongside other trypanosomes from cervids (North American elk and European fallow, red and sika deer), and bovids (cattle, antelopes and sheep). Insights into the life-cycle of T. trinaper- ronei n. sp. were obtained from early haemocultures of deer blood and co-culture with mammalian and insect cells showing fagellates resembling Megatrypanum trypanosomes previously reported in deer blood, and deer ked guts.