Ch 6 Polymer Materials and Their Technology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vinyl Chloride
Withdrawn Provided for Historical Reference Only VINYL CHLORIDE Method no.: 04 Matrix: Air Target concentration: 1 ppm (2.5 mg/m3) (OSHA PEL) Procedure: Collection on charcoal (two-tubes in-series) , desorption with carbon disulfide, analysis by gas chromatography with a flame ionization detector. Stored in refrigerator and analyzed as soon as possible. Detection limit based on recommended air volume: 0.25 ppm Recommended air volume and sampling rate: 1 L at 0.05 L/min Standard error of estimate at the target concentration: 7.6% (Figure 4.1.) Status of method: Recommended by NIOSH, partially evaluated by OSHA Laboratory. Date:WITHDRAWN April 1979 Chemist: Dee R. Chambers Organic Methods Evaluation Branch OSHA Analytical Laboratory Salt Lake City Utah Note: OSHA no longer uses or supports this method (December 2019). Withdrawn Provided for Historical Reference Only 1. General Discussion 1.1. Background 1.1.1. History In January 1974, B.F. Goodrich Chemical Company informed NIOSH of several deaths among polyvinyl chloride production workers from angiosarcoma, a rare liver cancer. In response to this and other evidence of potential hazards, OSHA lowered the workplace air standard for vinyl chloride (VC) from 500 ppm to 1 ppm 8-h time weighted average. Since the recognition that VC was a carcinogen, a great deal of research into the sampling and analytical methodology has been conducted. Perhaps the most complete and thorough examination has been conducted by Hill, McCammon, Saalwaechter, Teass, and W oodfin for NIOSH titled "The Gas Chromatographic Determination of Vinyl Chloride in Air Samples Collected on Charcoal" (Ref. 5.1.). -

2 Types of Noncrystalline Polymers
2 Types of Noncrystalline Polymers 1. Glassy polymer highly interpenetrated/entangled 2. Rubbery polymers random Gaussian coils Glass Transition Temperature εij Two viewpoints: • Increasing T: When kT > magnitude of εij, the thermal fluctuations can overcome local intermolecular bonds and the frozen (“glassy”) structure becomes “fluid-like”. • Decreasing T: As the temperature is lowered and T approaches Tg, the viscosity increases to ∞ and the material becomes “solid” Free Volume Theory of Tg Free volume, VF – extra space beyond what is present in an ordered crystalline packing (beyond the interstitial volume). VF(T) ≡ V(T) – V0(T) • V0 is occupied specific volume of atoms or molecules in the xline state and the spaces between them: ~ VXL. V(T) α • VF increases as T increases due to the Rate of cooling l difference in the thermal expansion V (T) αg XL coefficients (αg vs αl). FAST SLOW • V0(T) ≈ VXL(T) ↔ can take αg ≈αXL dV •V(T) = V (T ) + (T-T ) F T > T F F g g dT g Tg T • define fractional free volume, fF:Vf/V fF(T) = fF(Tg) + (T-Tg)αf αf = αl – αg Viewpoint: Tg occurs when available free volume drops below critical threshold for structural rearrangement [VITRIFICATION POINT], structure “jams up”. Tg Values of Amorphous Materials Table of representative amorphous solids, their bonding types, and their glass transition temperatures removed due to copyright restrictions. See Table 2.2 in Allen, S. M., and E.L. Thomas. The Structure of Materials. New York, NY: J. Wiley & Sons, 1999. Tg for Selected Polymers Glass Transition Temperature -

United States Patent (19) 11 Patent Number: 4,481,333 Fleischer Et Al
United States Patent (19) 11 Patent Number: 4,481,333 Fleischer et al. 45 Date of Patent: Nov. 6, 1984 54 THERMOPLASTIC COMPOSITIONS 58 Field of Search ................................ 525/192, 199 COMPRISING WINYL CHLORIDE POLYMER, CLPE AND FLUOROPOLYMER 56) References Cited U.S. PATENT DOCUMENTS 75) Inventors: Dietrich Fleischer, Darmstadt; Eckhard Weber, Liederbach; 3,005,795 10/1961 Busse et al. ......................... 525/199 3,294,871 2/1966 Schmitt et al. ...... 52.5/154 X Johannes Brandrup, Wiesbaden, all 3,299,182 1/1967 Jennings et al. ... ... 525/192 of Fed. Rep. of Germany 3,334,157 8/1967 Larsen ..................... ... 525/99 73 Assignee: Hoechst Aktiengesellschaft, Fed. 3,940,456 2/1976 Fey et al. ............................ 525/192 Rep. of Germany Primary Examiner-Carman J. Seccuro (21) Appl. No.: 566,207 Attorney, Agent, or Firm-Connolly & Hutz 22 Filed: Dec. 28, 1983 57 ABSTRACT 30 Foreign Application Priority Data The invention relates to a thermoplastic composition which comprises vinyl chloride polymers and chlori Dec. 31, 1982 (DE Fed. Rep. of Germany ....... 3248.731 nated polyethylene and which contains finely divided 51) Int. Cl. ...................... C08L 23/28; C08L 27/06; fluoropolymers and has a markedly improved process C08L 27/18 ability, particularly when shaped by extrusion. 52 U.S. C. .................................... 525/192; 525/199; 525/239 7 Claims, No Drawings 4,481,333 2 iaries and do not provide a solution to the present prob THERMOPLASTC COMPOSITIONS lem. COMPRISINGVINYL CHLORIDE POLYMER, -
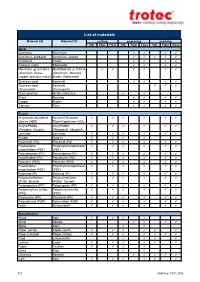
List of Materials
List of materials Material EN Material DE cutting engraving marking CO2 Fiber Flexx CO2 Fiber Flexx CO2 Fiber Flexx Metal Aluminum Aluminium Aluminum, anodized Aluminium, eloxiert Chromium Chrom Precious metal Edelmetall Metal foils up to 0.5mm Metallfolie bis zu 0,5mm (Aluminum, Brass, (Aluminium, Messing, Copper, precious metal) Kupfer, Edelmetall) Stainless steel Edelstahl Stainless steel Edelstahl (Thermark®) (Thermark®) Metal, painted Metall, lackiertes Brass Messing Copper Kupfer Titanium Titan Plastic Acrylonitrile butadiene Acrylnitril-Butadien- styrene (ABS) Styrol-Copolymer (ABS) Acrylic/PMMA Acryl/PMMA (Plexiglas, Altuglas, (Plexiglas®, Altuglas®, LaminatePerspex) LaminatePerspex®) Rubber Gummi Polyamide (PA) Polyamid (PA) Polybutylene Polybutylenterephthalat terephthalate (PBT) (PBT) Polycarbonate (PC) Polycarbonat (PC) Polyethylene (PE) Polyethylen (PE) Polyester (PES) Polyester (PES) Polyethylene Polyethylenterephthalat terephthalate (PET) (PET) Polyimide (PI) Polyimid (PI) Polyoxymethylene Polyoxymethylen (POM) -Delrin® (POM) - Delrin® Polypropylene (PP) Polypropylen (PP) Polyphenylene sulfide Polyphenylensulfid (PPS) (PPS) Polystyrene (PS) Polystyrol (PS) Polyurethane (PUR) Polyurethan (PUR) Foam Schaumstoff Miscellanious Wood Holz Mirror Spiegel Stone Stein Paper (white) Papier (weiß) Paper (colored) Papier (färbig) Food Lebensmittel Leather Leder -

Recent Attempts in the Design of Efficient PVC Plasticizers With
materials Review Recent Attempts in the Design of Efficient PVC Plasticizers with Reduced Migration Joanna Czogała 1,2,3,* , Ewa Pankalla 2 and Roman Turczyn 1,* 1 Department of Physical Chemistry and Technology of Polymers, Silesian University of Technology, Strzody 9, 44-100 Gliwice, Poland 2 Research and Innovation Department, Grupa Azoty Zakłady Azotowe K˛edzierzynS.A., Mostowa 30A, 47-220 K˛edzierzyn-Ko´zle,Poland; [email protected] 3 Joint Doctoral School, Silesian University of Technology, Akademicka 2A, 44-100 Gliwice, Poland * Correspondence: [email protected] (J.C.); [email protected] (R.T.) Abstract: This paper reviews the current trends in replacing commonly used plasticizers in poly(vinyl chloride), PVC, formulations by new compounds with reduced migration, leading to the enhance- ment in mechanical properties and better plasticizing efficiency. Novel plasticizers have been divided into three groups depending on the replacement strategy, i.e., total replacement, partial replacement, and internal plasticizers. Chemical and physical properties of PVC formulations containing a wide range of plasticizers have been compared, allowing observance of the improvements in polymer per- formance in comparison to PVC plasticized with conventionally applied bis(2-ethylhexyl) phthalate, di-n-octyl phthalate, bis(2-ethylhexyl) terephthalate and di-n-octyl terephthalate. Among a variety of newly developed plasticizers, we have indicated those presenting excellent migration resistance and advantageous mechanical properties, as well as those derived from natural sources. A separate chapter has been dedicated to the description of a synergistic effect of a mixture of two plasticizers, primary and secondary, that benefits in migration suppression when secondary plasticizer is added to PVC blend. -

Preventing Harm from Phthalates, Avoiding PVC in Hospitals
Preventing Harm from Phthalates, Avoiding PVC in Hospitals Karolina Ruzickova • Madeleine Cobbing • Mark Rossi • Thomas Belazzi Authors: Karolina Růžičková Madeleine Cobbing Mark Rossi, PhD. Thomas Belazzi, PhD. Acknowledgments: We would like to express our thanks to those who have contributed to the medical devices analysis and the content of the report: Prof. Ing. Bruno Klausbrückner, Vienna Hospital Association, Austria. Patricia Cameron and Friederike Otto, BUND – Friends of the Earth, Germany. Sonja Haider, Women in Europe for Common Future, Germany. Angelina Bartlett, Germany. Dorothee Lebeda, Germany. Aurélie Gauthier, CNIID, France. Lenka Mašková, Arnika, Czech Republic. Lumír Kantor, MD, Faculty Hospital Olomouc, Czech Republic. Juan Antonio Ortega García, Children’s Hospital La Fe, Spain. Malgorzata Kowalska, 3R, Poland. Anne Marie Vass, Karolinska University Hospital, Sweden. Åke Wennmalm, MD, PhD, Stockholm County Council, Sweden. Magnus Hedenmark, MSc., Sweden. We are deeply indebted to those have reviewed the report: Ted Schettler MD, MPH, Science and Environmental Health Network, USA. Charlotte Brody, RN and Stacy Malkan, Health Care Without Harm, USA. Sanford Lewis, Attorney, USA. And Dr.Čestmír Hrdinka, Health Care Without Harm, Czech Republic. We would also like to thank to Štěpán Bartošek and Štěpán Mamula for the design and production of the report and Stefan Gara for providing pictures. Preventing Harm from Phthalates, Avoiding PVC in Hospitals Heath Care Without Harm June 2004 Authors: Karolina Růžičková, Madeleine Cobbing, Mark Rossi, PhD., Thomas Belazzi, PhD. © 2004 Table of Contents Executive Summary ....................................................................................................................... 4 From Foetus to Toddler: Exposure to DEHP during a Critical Period of Development ........... 5 The Toxicity of DEHP DEHP Exposure during Specifi c Medical Treatment Therapies Results from Analytical Survey of Phthalates in Medical Products......................................... -

Photo-Stabilizers Utilizing Ibuprofen Tin Complexes Against Ultraviolet Radiation
Article A Surface Morphological Study, Poly(Vinyl Chloride) Photo-Stabilizers Utilizing Ibuprofen Tin Complexes against Ultraviolet Radiation Baraa Watheq 1, Emad Yousif 1,* , Mohammed H. Al-Mashhadani 1, Alaa Mohammed 1 , Dina S. Ahmed 2, Mohammed Kadhom 3 and Ali H. Jawad 4 1 Department of Chemistry, College of Science, Al-Nahrain University, 64021 Baghdad, Iraq; [email protected] (B.W.); [email protected] (M.H.A.-M.); [email protected] (A.M.) 2 Department of Medical Instrumentation Engineering, Al-Mansour University College, 64021 Baghdad, Iraq; [email protected] 3 Department of Renewable Energy, College of Energy and Environmental Science, Alkarkh University of Science, 64021 Baghdad, Iraq; [email protected] 4 Faculty of Applied Sciences, Universiti Teknologi MARA, Shah Alam 40450, Selangor, Malaysia; [email protected] * Correspondence: [email protected] Received: 23 August 2020; Accepted: 9 October 2020; Published: 13 October 2020 Abstract: In this work, three Ibuprofen tin complexes were synthesized and characterized by Fourier Transform Infrared spectroscopy (FTIR), 1H and 119Sn-Nuclear Magnetic Resonance (NMR), and Energy Dispersive X-ray (EDX) spectroscopies to identify the structures. The complexes were mixed separately with poly(vinyl chloride) (PVC) to improve its photo-stability properties. Their activity was demonstrated by several approaches of the FTIR to exhibit the formation of new groups within the polymer structure due to the exposure to UV light. Moreover, the polymer’s weight loss during irradiation and the average molecular weight estimation using its viscosity before and after irradiation were investigated. Furthermore, different techniques were used to study the surface morphology of the PVC before and after irradiation. -

Polyvinylchloride, Phthalates and Packaging
Page 29 | Bulletin 86 | July 2014 Polyvinylchloride, phthalates and packaging Plastics are synthetic resins, are either thermosetting or thermoplastic. Thermoplastic resins can be re-softened by heating, and include polyethylene (PE), polystyrene (PS), polypropylene (PP), and polyvinylchloride (PVC) which is very widely used in disposable medical devices. Polyvinylchloride (PVC) Di(2-ethylhexyl) phthalate (DEHP) Vinyl chloride (CH2=CHCl or chloroethylene) DEHP (sometimes referred to as bis is polymerized by free-radical initiators to (2-ethylhexyl) phthalate) is the diester of open the double bond and to link together phthalic acid and 2 ethylhexanol (Figure 2). vinyl chloride monomers, to form repeating Dr A M Walton units of polymers (Figure 1). Figure 2 The structure of di(2-ethylhexyl) phthalate DEHP. Anaesthesia ST 7, Figure 1 The phthalic moiety is common to all phthalates; University Hospital The chemical structure of vinyl chloride and PVC the pair of symmetrical aliphatic chains depends Southampton on the esterified alcohol PVC is a rigid structure at room temperature. Heating gives the molecules energy, widens the distance between between the molecules and softens the resin. To give PVC flexibility at room and body temperatures, plasticisers Dr J M T Pierce are added non-covalently to the PVC when Consultant Anaesthetist, At room temperature it is a colourless, oil- University Hospital molten and serve as molecular spacers so soluble viscous liquid and is used to form Southampton; RCoA when cooled the polymer has a softness even up to 40% of the mass of the PVC product. Environmental Advisor at room temperatures. The most commonly The annual global production of DEHP is used plasticisers are phthalates. -

Use of a Crosslinking Agent, Crosslinked Polymer, and Uses Thereof
(19) TZZ Z_T (11) EP 2 452 970 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C08K 5/09 (2006.01) B32B 27/30 (2006.01) 21.01.2015 Bulletin 2015/04 C08L 29/04 (2006.01) C08L 101/00 (2006.01) G02B 5/30 (2006.01) C08K 5/098 (2006.01) (2006.01) (21) Application number: 12154871.3 C08K 5/10 (22) Date of filing: 29.08.2008 (54) Use of a crosslinking agent, crosslinked polymer, and uses thereof Verwendung eines Vernetzungsmittels, vernetztes Polymer und Verwendungen davon Utilisation d’un agent de réticulation, polymère réticulé et utilisations associées (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Kageyama, Hideki HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT Osaka-shi, Osaka 530-0018 (JP) RO SE SI SK TR • Tanaka, Shinichi Osaka-shi, Osaka 530-0018 (JP) (30) Priority: 31.08.2007 JP 2007225012 • Ono, Hiroyuki 18.03.2008 JP 2008069645 Osaka-shi, Osaka 530-0018 (JP) 04.04.2008 JP 2008098282 • Kuruma, Akiko 11.04.2008 JP 2008103494 Osaka-shi, Osaka 530-0018 (JP) 24.04.2008 JP 2008114149 24.04.2008 JP 2008114150 (74) Representative: Kuhnen & Wacker Patent- und Rechtsanwaltsbüro (43) Date of publication of application: Prinz-Ludwig-Straße 40A 16.05.2012 Bulletin 2012/20 85354 Freising (DE) (62) Document number(s) of the earlier application(s) in (56) References cited: accordance with Art. 76 EPC: WO-A1-2005/087711 JP-B2- 3 612 124 08828502.8 / 2 189 496 US-A1- 2004 102 586 (73) Proprietor: The Nippon Synthetic Chemical • DATABASE WPI Week 198020 Thomson Industry Co., Ltd. -

BASEL CONVENTION PLASTICS AMENDMENTS Institute of Scrap Recycling Industries (ISRI)
BASEL CONVENTION PLASTICS AMENDMENTS Institute of Scrap Recycling Industries (ISRI) Adina Renee Adler Assistant Vice President, International Affairs ©2019 Institute of Scrap Recycling Industries, Inc. All Rights Reserved The Driver 2 Basel Convention Basel Convention on the Control of Transboundary Movements of Hazardous Wastes and Their Disposal Adopted 22 March 1989 Entered into force on 5 May 1992 Currently has 187 parties The United States is not a party. OCTOBER 2019 | ISRI.ORG 3 ©2019 ISRI, Inc. Basel Convention Basel Convention on the Control of Transboundary Movements of Hazardous Wastes and Their Disposal Subjects hazardous wastes or other wastes with hazardous constituents / exhibiting hazardous characteristics to a “prior informed consent” (PIC) procedure before the material can be shipped across national borders. The PIC gives the receiving country the right of refusal if proper recycling or disposal operations do not exist. OCTOBER 2019 | ISRI.ORG 4 ©2019 ISRI, Inc. Basel Convention 14th Conference of the Parties (COP) held every two years to direct work on implementing the convention and addressing emerging issues. OCTOBER 2019 | ISRI.ORG 5 ©2019 ISRI, Inc. Norway’s Proposal Amend the following annexes so that trade controls are 1 put into place for certain plastics: Annex II: Non-hazardous, hard-to-recycle plastics Annex VIII: Hazardous plastics Annex IX: Non-hazardous, easy-to-recycle plastics (exempt from BC) Create a Partnership for Plastic Waste that would involve public and private stakeholders to find possible solutions 2 to plastic leakage. Terms of Reference negotiated and adopted. It will lead to non- binding initiatives to support developing country improvements to minimization, collection and proper handling of end-of-life plastics. -

Vinyl Chloride CAS#: 75-01-4
PUBLIC HEALTH STATEMENT Vinyl Chloride CAS#: 75-01-4 Division of Toxicology and Environmental Medicine July 2006 This Public Health Statement is the summary breathing, eating, or drinking the substance, or by chapter from the Toxicological Profile for Vinyl skin contact. Chloride. It is one in a series of Public Health Statements about hazardous substances and their If you are exposed to vinyl chloride, many factors health effects. A shorter version, the ToxFAQs™, will determine whether you will be harmed. These is also available. This information is important factors include the dose (how much), the duration because this substance may harm you. The effects (how long), and how you come in contact with it. of exposure to any hazardous substance depend on You must also consider any other chemicals you are the dose, the duration, how you are exposed, exposed to and your age, sex, diet, family traits, personal traits and habits, and whether other lifestyle, and state of health. chemicals are present. For more information, call the ATSDR Information Center at 1-888-422-8737. 1.1 WHAT IS VINYL CHLORIDE? _____________________________________ Vinyl chloride is known also as chloroethene, This public health statement tells you about vinyl chloroethylene, ethylene monochloride, or chloride and the effects of exposure to it. monochloroethylene. At room temperature, it is a colorless gas, it burns easily, and it is not stable at The Environmental Protection Agency (EPA) high temperatures. Vinyl chloride exists in liquid identifies the most serious hazardous waste sites in form if kept under high pressure or at low the nation. These sites are then placed on the temperatures. -

State of Dibutyl Phthalate Molecules in Plasticized Poly(Vinyl Chloride)*
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Kazan Federal University Digital Repository Polymer Science U.S.S.R. Vol. 20, pp. 1492-1498. 0932-3950/78/9601-1492507.50]0. (~) Pergamon Press Ltd. 1979. Printed in Poland. STATE OF DIBUTYL PHTHALATE MOLECULES IN PLASTICIZED POLY(VINYL CHLORIDE)* A. ~. ~AkKLAKOV, A. A. I~¢[AKLAKOV, A. N. TEMlqIKOV and B. F. TEPLOV V. I. Lenin State University, Kazan V. A. Kargin Polymer Chemistry and Technology Research Institute J (Received 22 August 1977) It was found that the rotational motion of plasticizer molecules in the polymer matrix is hindered, the degree of hindrance differing for different molecules. With " use of the data on the dibutyl phthalate (DBP) autodiffusion it" was possible to evaluate the lifetime of the PVC-plasticizer solvates. This lifetime is a function of the amount of small molecules and of temperature. The solvatation in these systems is of the dynamic type. The "free" plasticizer is absent in samples containing 30-52 wt. °/o DBP. The measurements were made by the pulse NMR method. SEVERAL investigations have been reported in connection with the state of plasticizer molecules introduced into polymer [1]. However, the problem has yet to be fully resolved. In some cases use of the NMR method allows separate evalua- tion of the mobility of macromolecules and molecules of a low molecular substance and it appears that the use of NMR may provide additional information on the mode of behaviour of molecules of components forming part of plasticized sys- tems.