Theoretical Description of Magnetic Compton Scattering and Magnetic Properties of Cr-Chalcogenide Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Compton Scattering from the Deuteron at Low Energies
SE0200264 LUNFD6-NFFR-1018 Compton Scattering from the Deuteron at Low Energies Magnus Lundin Department of Physics Lund University 2002 W' •sii" Compton spridning från deuteronen vid låga energier (populärvetenskaplig sammanfattning på svenska) Vid Compton spridning sprids fotonen elastiskt, dvs. utan att förlora energi, mot en annan partikel eller kärna. Kärnorna som användes i detta försök består av en proton och en neutron (sk. deuterium, eller tungt väte). Kärnorna bestrålades med fotoner av kända energier och de spridda fo- tonerna detekterades mha. stora Nal-detektorer som var placerade i olika vinklar runt strålmålet. Försöket utfördes under 8 veckor och genom att räkna antalet fotoner som kärnorna bestålades med och antalet spridda fo- toner i de olika detektorerna, kan sannolikheten för att en foton skall spridas bestämmas. Denna sannolikhet jämfördes med en teoretisk modell som beskriver sannolikheten för att en foton skall spridas elastiskt mot en deuterium- kärna. Eftersom protonen och neutronen består av kvarkar, vilka har en elektrisk laddning, kommer dessa att sträckas ut då de utsätts för ett elek- triskt fält (fotonen), dvs. de polariseras. Värdet (sannolikheten) som den teoretiska modellen ger, beror på polariserbarheten hos protonen och neu- tronen i deuterium. Genom att beräkna sannolikheten för fotonspridning för olika värden av polariserbarheterna, kan man se vilket värde som ger bäst överensstämmelse mellan modellen och experimentella data. Det är speciellt neutronens polariserbarhet som är av intresse, och denna kunde bestämmas i detta arbete. Organization Document name LUND UNIVERSITY DOCTORAL DISSERTATION Department of Physics Date of issue 2002.04.29 Division of Nuclear Physics Box 118 Sponsoring organization SE-22100 Lund Sweden Author (s) Magnus Lundin Title and subtitle Compton Scattering from the Deuteron at Low Energies Abstract A series of three Compton scattering experiments on deuterium have been performed at the high-resolution tagged-photon facility MAX-lab located in Lund, Sweden. -

7. Gamma and X-Ray Interactions in Matter
Photon interactions in matter Gamma- and X-Ray • Compton effect • Photoelectric effect Interactions in Matter • Pair production • Rayleigh (coherent) scattering Chapter 7 • Photonuclear interactions F.A. Attix, Introduction to Radiological Kinematics Physics and Radiation Dosimetry Interaction cross sections Energy-transfer cross sections Mass attenuation coefficients 1 2 Compton interaction A.H. Compton • Inelastic photon scattering by an electron • Arthur Holly Compton (September 10, 1892 – March 15, 1962) • Main assumption: the electron struck by the • Received Nobel prize in physics 1927 for incoming photon is unbound and stationary his discovery of the Compton effect – The largest contribution from binding is under • Was a key figure in the Manhattan Project, condition of high Z, low energy and creation of first nuclear reactor, which went critical in December 1942 – Under these conditions photoelectric effect is dominant Born and buried in • Consider two aspects: kinematics and cross Wooster, OH http://en.wikipedia.org/wiki/Arthur_Compton sections http://www.findagrave.com/cgi-bin/fg.cgi?page=gr&GRid=22551 3 4 Compton interaction: Kinematics Compton interaction: Kinematics • An earlier theory of -ray scattering by Thomson, based on observations only at low energies, predicted that the scattered photon should always have the same energy as the incident one, regardless of h or • The failure of the Thomson theory to describe high-energy photon scattering necessitated the • Inelastic collision • After the collision the electron departs -
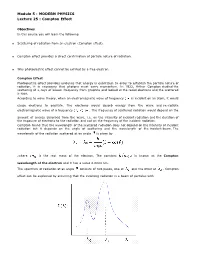
Compton Effect
Module 5 : MODERN PHYSICS Lecture 25 : Compton Effect Objectives In this course you will learn the following Scattering of radiation from an electron (Compton effect). Compton effect provides a direct confirmation of particle nature of radiation. Why photoelectric effect cannot be exhited by a free electron. Compton Effect Photoelectric effect provides evidence that energy is quantized. In order to establish the particle nature of radiation, it is necessary that photons must carry momentum. In 1922, Arthur Compton studied the scattering of x-rays of known frequency from graphite and looked at the recoil electrons and the scattered x-rays. According to wave theory, when an electromagnetic wave of frequency is incident on an atom, it would cause electrons to oscillate. The electrons would absorb energy from the wave and re-radiate electromagnetic wave of a frequency . The frequency of scattered radiation would depend on the amount of energy absorbed from the wave, i.e. on the intensity of incident radiation and the duration of the exposure of electrons to the radiation and not on the frequency of the incident radiation. Compton found that the wavelength of the scattered radiation does not depend on the intensity of incident radiation but it depends on the angle of scattering and the wavelength of the incident beam. The wavelength of the radiation scattered at an angle is given by .where is the rest mass of the electron. The constant is known as the Compton wavelength of the electron and it has a value 0.0024 nm. The spectrum of radiation at an angle consists of two peaks, one at and the other at . -

Compton Scattering from Low to High Energies
Compton Scattering from Low to High Energies Marc Vanderhaeghen College of William & Mary / JLab HUGS 2004 @ JLab, June 1-18 2004 Outline Lecture 1 : Real Compton scattering on the nucleon and sum rules Lecture 2 : Forward virtual Compton scattering & nucleon structure functions Lecture 3 : Deeply virtual Compton scattering & generalized parton distributions Lecture 4 : Two-photon exchange physics in elastic electron-nucleon scattering …if you want to read more details in preparing these lectures, I have primarily used some review papers : Lecture 1, 2 : Drechsel, Pasquini, Vdh : Physics Reports 378 (2003) 99 - 205 Lecture 3 : Guichon, Vdh : Prog. Part. Nucl. Phys. 41 (1998) 125 – 190 Goeke, Polyakov, Vdh : Prog. Part. Nucl. Phys. 47 (2001) 401 - 515 Lecture 4 : research papers , field in rapid development since 2002 1st lecture : Real Compton scattering on the nucleon & sum rules IntroductionIntroduction :: thethe realreal ComptonCompton scatteringscattering (RCS)(RCS) processprocess ε, ε’ : photon polarization vectors σ, σ’ : nucleon spin projections Kinematics in LAB system : shift in wavelength of scattered photon Compton (1923) ComptonCompton scatteringscattering onon pointpoint particlesparticles Compton scattering on spin 1/2 point particle (Dirac) e.g. e- Klein-Nishina (1929) : Thomson term ΘL = 0 Compton scattering on spin 1/2 particle with anomalous magnetic moment Stern (1933) Powell (1949) LowLow energyenergy expansionexpansion ofof RCSRCS processprocess Spin-independent RCS amplitude note : transverse photons : Low -

The Basic Interactions Between Photons and Charged Particles With
Outline Chapter 6 The Basic Interactions between • Photon interactions Photons and Charged Particles – Photoelectric effect – Compton scattering with Matter – Pair productions Radiation Dosimetry I – Coherent scattering • Charged particle interactions – Stopping power and range Text: H.E Johns and J.R. Cunningham, The – Bremsstrahlung interaction th physics of radiology, 4 ed. – Bragg peak http://www.utoledo.edu/med/depts/radther Photon interactions Photoelectric effect • Collision between a photon and an • With energy deposition atom results in ejection of a bound – Photoelectric effect electron – Compton scattering • The photon disappears and is replaced by an electron ejected from the atom • No energy deposition in classical Thomson treatment with kinetic energy KE = hν − Eb – Pair production (above the threshold of 1.02 MeV) • Highest probability if the photon – Photo-nuclear interactions for higher energies energy is just above the binding energy (above 10 MeV) of the electron (absorption edge) • Additional energy may be deposited • Without energy deposition locally by Auger electrons and/or – Coherent scattering Photoelectric mass attenuation coefficients fluorescence photons of lead and soft tissue as a function of photon energy. K and L-absorption edges are shown for lead Thomson scattering Photoelectric effect (classical treatment) • Electron tends to be ejected • Elastic scattering of photon (EM wave) on free electron o at 90 for low energy • Electron is accelerated by EM wave and radiates a wave photons, and approaching • No -

Interactions of Photons with Matter
22.55 “Principles of Radiation Interactions” Interactions of Photons with Matter • Photons are electromagnetic radiation with zero mass, zero charge, and a velocity that is always c, the speed of light. • Because they are electrically neutral, they do not steadily lose energy via coulombic interactions with atomic electrons, as do charged particles. • Photons travel some considerable distance before undergoing a more “catastrophic” interaction leading to partial or total transfer of the photon energy to electron energy. • These electrons will ultimately deposit their energy in the medium. • Photons are far more penetrating than charged particles of similar energy. Energy Loss Mechanisms • photoelectric effect • Compton scattering • pair production Interaction probability • linear attenuation coefficient, µ, The probability of an interaction per unit distance traveled Dimensions of inverse length (eg. cm-1). −µ x N = N0 e • The coefficient µ depends on photon energy and on the material being traversed. µ • mass attenuation coefficient, ρ The probability of an interaction per g cm-2 of material traversed. Units of cm2 g-1 ⎛ µ ⎞ −⎜ ⎟()ρ x ⎝ ρ ⎠ N = N0 e Radiation Interactions: photons Page 1 of 13 22.55 “Principles of Radiation Interactions” Mechanisms of Energy Loss: Photoelectric Effect • In the photoelectric absorption process, a photon undergoes an interaction with an absorber atom in which the photon completely disappears. • In its place, an energetic photoelectron is ejected from one of the bound shells of the atom. • For gamma rays of sufficient energy, the most probable origin of the photoelectron is the most tightly bound or K shell of the atom. • The photoelectron appears with an energy given by Ee- = hv – Eb (Eb represents the binding energy of the photoelectron in its original shell) Thus for gamma-ray energies of more than a few hundred keV, the photoelectron carries off the majority of the original photon energy. -

Fundamental Study of Compton Scattering Tomography
FUNDAMENTAL STUDY OF COMPTON SCATTERING TOMOGRAPHY R. XIAO, S. SATO, C. UYAMAf, and T. NAKAMURAJ Department of Biophysical Engineering, Faculty of Engineering Science, Osaka University \ Department of Investigative Radiology, National Cardiovascular Center Research Institute \Cyclotron and Radioisotope Center, Tohoku University 1. Introduction Compton scattering tomography is an imaging technique that forms tomographic im ages corresponding to distribution of electron densities in an X-rays or 7-rays1 irradiating object by measuring Compton scattered photons emitted out of it. Because of its advan tage of free choice of measurement geometry, Compton scattering tomography is useful for inspection of defects inside huge objects in industry where X-rays or 7-rays can hardly pen etrate the objects[l]. For medical usage, Compton scattered rays are used for densitometry of bone or other tissues. Compton scattering tomographic images, in which densities of body other than attenuation coefficients of body can be shown, are meaningful for medical diagnoses. Before building a real Compton scattering tomography system for medical applica tions, we made some fundamental observations upon Compton scattering imaging process through Monte Carlo simulation in order to testify feasibility of the imaging and to seek for an available scheme of system design. EGS4 code system[2] is used for simulation of transport processes of photons in an imaged object. Multiple scattering, one of factors that influence quantity of Compton scattering images, is evaluated here. Some methods to overcome multiple scattering are proposed and their effects are discussed below. 2. Principle of Compton scattering tomography A beam of X-rays or 7-rays attenuates in its intensity by interactions of photoelectric absorption, Compton scattering, Rayleigh scattering and electron-positron pair production while it is passing through an object. -

Lecture 18 Quantum Electrodynamics
Lecture 18 Quantum Electrodynamics Quantum Electrodynamics (QED) describes a U(1) gauge theory coupled to a fermion, e.g. the electron, muon or any other charged particle. As we saw previously the lagrangian for such theory is 1 L = ¯(iD6 − m) − F F µν ; (18.1) 4 µν where the covariant derivative is defined as Dµ (x) = (@µ + ieAµ(x)) (x) ; (18.2) resulting in 1 L = ¯(i6@ − m) − F F µν − eA ¯γµ ; (18.3) 4 µν µ where the last term is the interaction lagrangian and thus e is the photon-fermion cou- pling. We would like to derive the Feynman rules for this theory, and then compute the amplitudes and cross sections for some interesting processes. 18.1 Feynman Rules for QED In addition to the rules for the photon and fermion propagator, we now need to derive the Feynman rule for the photon-fermion vertex. One way of doing this is to use the generating functional in presence of linearly coupled external sources Z ¯ R 4 µ ¯ ¯ iS[ ; ;Aµ]+i d xfJµA +¯η + ηg Z[η; η;¯ Jµ] = N D D DAµ e : (18.4) 1 2 LECTURE 18. QUANTUM ELECTRODYNAMICS From it we can derive a three-point function with an external photon, a fermion and an antifermion. This is given by 3 3 (3) (−i) δ Z[η; η;¯ Jµ] G (x1; x2; x3) = ; (18.5) Z[0; 0; 0] δJ (x )δη(x )δη¯(x ) µ 1 2 3 Jµ,η;η¯=0 which, when we expand in the interaction term, results in 1 Z ¯ G(3)(x ; x ; x ) = D ¯ D DA eiS0[ ; ;Aµ]A (x ) ¯(x ) (x ) 1 2 3 Z[0; 0; 0] µ µ 1 2 3 Z 4 ¯ ν × 1 − ie d yAν(y) (y)γ (y) + ::: Z 4 ν = (+ie) d yDF µν(x1 − y)Tr [SF(x3 − y)γ SF(x2 − y)] ; (18.6) where S0 is the free action, obtained from the first two terms in (18.3). -

Electron Scattering Processes in Non-Monochromatic and Relativistically Intense Laser Fields
atoms Review Electron Scattering Processes in Non-Monochromatic and Relativistically Intense Laser Fields Felipe Cajiao Vélez * , Jerzy Z. Kami ´nskiand Katarzyna Krajewska Institute of Theoretical Physics, Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland; [email protected] (J.Z.K.); [email protected] (K.K.) * Correspondence: [email protected]; Tel.: +48-22-5532920 Received: 31 January 2019; Accepted: 1 March 2019; Published: 6 March 2019 Abstract: The theoretical analysis of four fundamental laser-assisted non-linear scattering processes are summarized in this review. Our attention is focused on Thomson, Compton, Møller and Mott scattering in the presence of intense electromagnetic radiation. Depending on the phenomena under considerations, we model the laser field as a single laser pulse of ultrashort duration (for Thomson and Compton scattering) or non-monochromatic trains of pulses (for Møller and Mott scattering). Keywords: laser-assisted quantum processes; non-linear scattering; ultrashort laser pulses 1. Introduction Scattering theory is the part of theoretical physics in which interactions of particles and waves are investigated at remote times and large distances, as compared with typical time and size scales of probed systems. For this reason, scattering theory is the most effective and, in many cases, the only method of analyzing such diverse systems as the micro-world or the whole universe. Not surprisingly, the investigation of scattering phenomena has been playing a central role in physics since the end of the nineteenth century, starting from the Rayleigh’s explanation of why the sky is blue, up to modern medical applications of computerized tomography, the very recent discovery of the Higgs particle and the detection of gravitational waves. -

Probing Entanglement in Compton Interactions Arxiv:1910.05537V1
P. Caradonna et al. Probing entanglement in Compton interactions Peter Caradonna1∗, David Reutens1;2, Tadayuki Takahashi3, Shin'ichiro Takeda3, Viktor Vegh1;2 1Centre for Advanced Imaging, The University of Queensland, Brisbane 4072, Australia 2ARC Training Centre for Innovation in Biomedical Imaging Technology, Australia 3Kavli Institute for the Physics and Mathematics of the Universe (WPI), Institutes for Advanced Study (UTIAS), The University of Tokyo, 5-1-5 Kashiwa-no-Ha, Kashiwa, Chiba, 277-8583, Japan ∗ Present address: Kavli Institute for the Physics and Mathematics of the Universe (WPI), In- stitutes for Advanced Study (UTIAS), The University of Tokyo, 5-1-5 Kashiwa-no-Ha, Kashiwa, Chiba, 277-8583, Japan Email: [email protected] Keywords: Entanglement, Compton scattering, Bell States, Compton cameras, Compton PET Abstract This theoretical research aims to examine areas of the Compton cross section of entangled anni- hilation photons for the purpose of testing for possible break down of theory, which could have consequences for predicted optimal capabilities of Compton PET systems.We provide maps of the cross section for entangled annihilation photons for experimental verification.We introduce a strat- egy to derive cross sections in a relatively straight forward manner for the Compton scattering of a hypothetical separable, mixed and entangled states. To understand the effect that entanglement has on the cross section for annihilation photons, we derive the cross section so that it is expressed in terms of the cross section -

Compton Scattering 1 Compton Scattering
Compton scattering 1 Compton scattering Light–matter interaction Low-energy phenomena: Photoelectric effect Mid-energy phenomena: Thomson scattering Compton scattering High-energy phenomena: Pair production • v • t [1] • e Compton scattering is an inelastic scattering of a photon by a free charged particle, usually an electron. It results in a decrease in energy (increase in wavelength) of the photon (which may be an X-ray or gamma ray photon), called the Compton effect. Part of the energy of the photon is transferred to the recoiling electron. Inverse Compton scattering also exists, in which a charged particle transfers part of its energy to a photon. Introduction Compton scattering is an example of inelastic scattering, because the wavelength of the scattered light is different from the incident radiation. Still, the origin of the effect can be considered as an elastic collision between a photon and an electron. The amount the wavelength changes by is called the Compton shift. Although nuclear Compton scattering exists, Compton scattering usually refers to the interaction involving only the electrons of an atom. The Compton effect was observed by Arthur Holly Compton in 1923 at Washington University in St. Louis and further verified by his graduate student Y. H. Woo in the years following. Compton earned the 1927 Nobel Prize in Physics for the discovery. The effect is important because it demonstrates that light cannot be explained purely as a wave phenomenon. Thomson scattering, the classical theory of an electromagnetic wave scattered by charged particles, cannot explain low intensity shifts in wavelength. (Classically, light of sufficient intensity for the electric field to accelerate a charged particle to a relativistic speed will cause radiation-pressure recoil and an associated Doppler shift of the scattered light,[2] but the effect would become arbitrarily small at sufficiently low light intensities regardless of wavelength.) Light must behave as if it consists of particles to explain the low-intensity Compton scattering. -

Basic Health Physics
Interaction of Photons With Matter 1 7/5/2011 General Stuff 2 Objectives • To review three principal interactions of photons with matter. • To examine the factors affecting the probability of photon interactions. • To introduce the linear and mass attenuation coefficients as useful measures of the probability of interaction. 3 Introduction • Understanding how radiation interacts with matter is essential to understanding: – How instruments detect radiation – How dose is delivered to tissue – How to design an effective shield 4 Properties of Photons • A photon is a “packet” of electromagnetic energy. • It has no mass, no charge, and travels in a straight line at the speed of light. 5 6 Properties of Photons (cont'd) This discussion is limited to photons with enough energy to ionize matter: • X-rays: characteristic x-rays bremsstrahlung • gamma rays • annihilation radiation (511 keV) 7 Photon Interactions • Photons interact differently in matter than charged particles because photons have no electrical charge. • In contrast to charged particles, photons do not continuously lose energy when they travel through matter. 8 Photon Interactions (cont'd) • When photons interact, they transfer energy to charged particles (usually electrons) and the charged particles githiiive up their energy via secon dary interactions (mostly ionization). • The interaction of photons with matter is probabilistic, while the interaction of charged particles is certain. 9 +++++ ------- alpha - + e- Beta Possible Gamma Interactions 10 Gamma e- 11 Gamma ? 12 Photon Interactions (cont'd) • The probability of a photon interacting depends upon: – The photon energy – The atomic number and density of the material (electron density of the absorbing matter). 13 Photon Interactions (cont'd) • The three principal types of photon interactions are: – Phot oel ect ri c eff ec t – Compton scattering (incoherent scattering) – Pair production 14 Photon Interactions (cont'd) • There are a number of less important mechanisms by which photons interact with matter.