GABRB3 Mutations: a New and Emerging Cause of Early Infantile Epileptic Encephalopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Coupling of Autism Genes to Tissue-Wide Expression and Dysfunction of Synapse, Calcium Signalling and Transcriptional Regulation
PLOS ONE RESEARCH ARTICLE Coupling of autism genes to tissue-wide expression and dysfunction of synapse, calcium signalling and transcriptional regulation 1 2,3 4 1,5 Jamie ReillyID *, Louise Gallagher , Geraldine Leader , Sanbing Shen * 1 Regenerative Medicine Institute, School of Medicine, Biomedical Science Building, National University of a1111111111 Ireland (NUI) Galway, Galway, Ireland, 2 Discipline of Psychiatry, School of Medicine, Trinity College Dublin, Dublin, Ireland, 3 Trinity Translational Medicine Institute, Trinity Centre for Health SciencesÐTrinity College a1111111111 Dublin, St. James's Hospital, Dublin, Ireland, 4 Irish Centre for Autism and Neurodevelopmental Research a1111111111 (ICAN), Department of Psychology, National University of Ireland (NUI) Galway, Galway, Ireland, a1111111111 5 FutureNeuro Research Centre, Royal College of Surgeons in Ireland (RCSI), Dublin, Ireland a1111111111 * [email protected] (JR); [email protected] (SS) Abstract OPEN ACCESS Citation: Reilly J, Gallagher L, Leader G, Shen S Autism Spectrum Disorder (ASD) is a heterogeneous disorder that is often accompanied (2020) Coupling of autism genes to tissue-wide with many co-morbidities. Recent genetic studies have identified various pathways from expression and dysfunction of synapse, calcium hundreds of candidate risk genes with varying levels of association to ASD. However, it is signalling and transcriptional regulation. PLoS ONE unknown which pathways are specific to the core symptoms or which are shared by the co- 15(12): e0242773. https://doi.org/10.1371/journal. pone.0242773 morbidities. We hypothesised that critical ASD candidates should appear widely across dif- ferent scoring systems, and that comorbidity pathways should be constituted by genes Editor: Nirakar Sahoo, The University of Texas Rio Grande Valley, UNITED STATES expressed in the relevant tissues. -

Mechanisms Underlying the EEG Biomarker in Dup15q Syndrome Joel Frohlich1,2,3* , Lawrence T
Frohlich et al. Molecular Autism (2019) 10:29 https://doi.org/10.1186/s13229-019-0280-6 RESEARCH Open Access Mechanisms underlying the EEG biomarker in Dup15q syndrome Joel Frohlich1,2,3* , Lawrence T. Reiter4, Vidya Saravanapandian2, Charlotte DiStefano2, Scott Huberty2,5, Carly Hyde2, Stormy Chamberlain6, Carrie E. Bearden7, Peyman Golshani8, Andrei Irimia9, Richard W. Olsen10, Joerg F. Hipp1† and Shafali S. Jeste2† Abstract Background: Duplications of 15q11.2-q13.1 (Dup15q syndrome), including the paternally imprinted gene UBE3A and three nonimprinted gamma-aminobutyric acid type-A (GABAA) receptor genes, are highly penetrant for neurodevelopmental disorders such as autism spectrum disorder (ASD). To guide targeted treatments of Dup15q syndrome and other forms of ASD, biomarkers are needed that reflect molecular mechanisms of pathology. We recently described a beta EEG phenotype of Dup15q syndrome, but it remains unknown which specific genes drive this phenotype. Methods: To test the hypothesis that UBE3A overexpression is not necessary for the beta EEG phenotype, we compared EEG from a reference cohort of children with Dup15q syndrome (n = 27) to (1) the pharmacological effects of the GABAA modulator midazolam (n = 12) on EEG from healthy adults, (2) EEG from typically developing (TD) children (n = 14), and (3) EEG from two children with duplications of paternal 15q (i.e., the UBE3A-silenced allele). Results: Peak beta power was significantly increased in the reference cohort relative to TD controls. Midazolam administration recapitulated the beta EEG phenotype in healthy adults with a similar peak frequency in central channels (f = 23.0 Hz) as Dup15q syndrome (f = 23.1 Hz). -

Characterization of Zebrafish GABAA Receptor Subunits
www.nature.com/scientificreports OPEN Characterization of zebrafsh GABAA receptor subunits Kenichiro Sadamitsu, Leona Shigemitsu, Marina Suzuki, Daishi Ito, Makoto Kashima & Hiromi Hirata* γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system, exerts its efect through the activation of GABA receptors. GABAA receptors are ligand-gated chloride channels composed of fve subunit proteins. Mammals have 19 diferent GABAA receptor subunits (α1–6, β1–3, γ1–3, δ, ε, π, θ, and ρ1–3), the physiological properties of which have been assayed by electrophysiology. However, the evolutionary conservation of the physiological characteristics of diverged GABAA receptor subunits remains unclear. Zebrafsh have 23 subunits (α1, α2a, α2b, α3–5, α6a, α6b, β1–4, γ1–3, δ, π, ζ, ρ1, ρ2a, ρ2b, ρ3a, and ρ3b), but the electrophysiological properties of these subunits have not been explored. In this study, we cloned the coding sequences for zebrafsh GABAA receptor subunits and investigated their expression patterns in larval zebrafsh by whole- mount in situ hybridization. We also performed electrophysiological recordings of GABA-evoked currents from Xenopus oocytes injected with one or multiple zebrafsh GABAA receptor subunit cRNAs and calculated the half-maximal efective concentrations (EC50s) for each. Our results revealed the spatial expressions and electrophysiological GABA sensitivities of zebrafsh GABAA receptors, suggesting that the properties of GABAA receptor subunits are conserved among vertebrates. γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system of vertebrates, 1 controls the excitability of neural networks mainly through GABA A receptors . Te GABAA receptor mediates two types of inhibition, known as phasic and tonic inhibition2. -

Supplementary Material
Supplementary Material Table S1: Significant downregulated KEGGs pathways identified by DAVID following exposure to five cinnamon- based phenylpropanoids (p < 0.05). p-value Term: Genes (Benjamini) Cytokine-cytokine receptor interaction: FASLG, TNFSF14, CXCL11, IL11, FLT3LG, CCL3L1, CCL3L3, CXCR6, XCR1, 2.43 × 105 RTEL1, CSF2RA, TNFRSF17, TNFRSF14, CCNL2, VEGFB, AMH, TNFRSF10B, INHBE, IFNB1, CCR3, VEGFA, CCR2, IL12A, CCL1, CCL3, CXCL5, TNFRSF25, CCR1, CSF1, CX3CL1, CCL7, CCL24, TNFRSF1B, IL12RB1, CCL21, FIGF, EPO, IL4, IL18R1, FLT1, TGFBR1, EDA2R, HGF, TNFSF8, KDR, LEP, GH2, CCL13, EPOR, XCL1, IFNA16, XCL2 Neuroactive ligand-receptor interaction: OPRM1, THRA, GRIK1, DRD2, GRIK2, TACR2, TACR1, GABRB1, LPAR4, 9.68 × 105 GRIK5, FPR1, PRSS1, GNRHR, FPR2, EDNRA, AGTR2, LTB4R, PRSS2, CNR1, S1PR4, CALCRL, TAAR5, GABRE, PTGER1, GABRG3, C5AR1, PTGER3, PTGER4, GABRA6, GABRA5, GRM1, PLG, LEP, CRHR1, GH2, GRM3, SSTR2, Chlorogenic acid Chlorogenic CHRM3, GRIA1, MC2R, P2RX2, TBXA2R, GHSR, HTR2C, TSHR, LHB, GLP1R, OPRD1 Hematopoietic cell lineage: IL4, CR1, CD8B, CSF1, FCER2, GYPA, ITGA2, IL11, GP9, FLT3LG, CD38, CD19, DNTT, 9.29 × 104 GP1BB, CD22, EPOR, CSF2RA, CD14, THPO, EPO, HLA-DRA, ITGA2B Cytokine-cytokine receptor interaction: IL6ST, IL21R, IL19, TNFSF15, CXCR3, IL15, CXCL11, TGFB1, IL11, FLT3LG, CXCL10, CCR10, XCR1, RTEL1, CSF2RA, IL21, CCNL2, VEGFB, CCR8, AMH, TNFRSF10C, IFNB1, PDGFRA, EDA, CXCL5, TNFRSF25, CSF1, IFNW1, CNTFR, CX3CL1, CCL5, TNFRSF4, CCL4, CCL27, CCL24, CCL25, CCL23, IFNA6, IFNA5, FIGF, EPO, AMHR2, IL2RA, FLT4, TGFBR2, EDA2R, -

Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed Hipscs To
Hindawi Publishing Corporation Stem Cells International Volume 2013, Article ID 784629, 25 pages http://dx.doi.org/10.1155/2013/784629 Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed hiPSCs to Differentiated Neuronal and Cardiac Progeny Leonhard Linta,1 Marianne Stockmann,1 Qiong Lin,2 André Lechel,3 Christian Proepper,1 Tobias M. Boeckers,1 Alexander Kleger,3 and Stefan Liebau1 1 InstituteforAnatomyCellBiology,UlmUniversity,Albert-EinsteinAllee11,89081Ulm,Germany 2 Institute for Biomedical Engineering, Department of Cell Biology, RWTH Aachen, Pauwelstrasse 30, 52074 Aachen, Germany 3 Department of Internal Medicine I, Ulm University, Albert-Einstein Allee 11, 89081 Ulm, Germany Correspondence should be addressed to Alexander Kleger; [email protected] and Stefan Liebau; [email protected] Received 31 January 2013; Accepted 6 March 2013 Academic Editor: Michael Levin Copyright © 2013 Leonhard Linta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Ion channels are involved in a large variety of cellular processes including stem cell differentiation. Numerous families of ion channels are present in the organism which can be distinguished by means of, for example, ion selectivity, gating mechanism, composition, or cell biological function. To characterize the distinct expression of this group of ion channels we have compared the mRNA expression levels of ion channel genes between human keratinocyte-derived induced pluripotent stem cells (hiPSCs) and their somatic cell source, keratinocytes from plucked human hair. This comparison revealed that 26% of the analyzed probes showed an upregulation of ion channels in hiPSCs while just 6% were downregulated. -

Assessment of Molecular Action of Direct Gating and Allosteric Modulatory Effects of Carisoprodol (Somartm) on GABA a Receptors
Graduate Theses, Dissertations, and Problem Reports 2015 Assessment of molecular action of direct gating and allosteric modulatory effects of carisoprodol (SomaRTM) on GABA A receptors Manoj Kumar Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Kumar, Manoj, "Assessment of molecular action of direct gating and allosteric modulatory effects of carisoprodol (SomaRTM) on GABA A receptors" (2015). Graduate Theses, Dissertations, and Problem Reports. 6022. https://researchrepository.wvu.edu/etd/6022 This Dissertation is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. ASSESSMENT OF MOLECULAR ACTION OF DIRECT GATING AND ALLOSTERIC MODULATORY EFFECTS OF MEPROBAMATE (MILTOWN®) ON GABAA RECEPTORS Manish Kumar, MD, MS Dissertation submitted to the School of Pharmacy at West Virginia University in partial fulfillment of Requirements -

Identification of Key Genes and Pathways Involved in Response To
Deng et al. Biol Res (2018) 51:25 https://doi.org/10.1186/s40659-018-0174-7 Biological Research RESEARCH ARTICLE Open Access Identifcation of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing Xiuling Deng1,2†, Dong Wang3†, Shenyuan Wang1, Haisheng Wang2 and Huanmin Zhou1* Abstract Purpose: This aim of this study was to investigate the key genes and pathways involved in the response to pain in goat and sheep by transcriptome sequencing. Methods: Chronic pain was induced with the injection of the complete Freund’s adjuvant (CFA) in sheep and goats. The animals were divided into four groups: CFA-treated sheep, control sheep, CFA-treated goat, and control goat groups (n 3 in each group). The dorsal root ganglions of these animals were isolated and used for the construction of a cDNA= library and transcriptome sequencing. Diferentially expressed genes (DEGs) were identifed in CFA-induced sheep and goats and gene ontology (GO) enrichment analysis was performed. Results: In total, 1748 and 2441 DEGs were identifed in CFA-treated goat and sheep, respectively. The DEGs identi- fed in CFA-treated goats, such as C-C motif chemokine ligand 27 (CCL27), glutamate receptor 2 (GRIA2), and sodium voltage-gated channel alpha subunit 3 (SCN3A), were mainly enriched in GO functions associated with N-methyl- D-aspartate (NMDA) receptor, infammatory response, and immune response. The DEGs identifed in CFA-treated sheep, such as gamma-aminobutyric acid (GABA)-related DEGs (gamma-aminobutyric acid type A receptor gamma 3 subunit [GABRG3], GABRB2, and GABRB1), SCN9A, and transient receptor potential cation channel subfamily V member 1 (TRPV1), were mainly enriched in GO functions related to neuroactive ligand-receptor interaction, NMDA receptor, and defense response. -

Stem Cells and Ion Channels
Stem Cells International Stem Cells and Ion Channels Guest Editors: Stefan Liebau, Alexander Kleger, Michael Levin, and Shan Ping Yu Stem Cells and Ion Channels Stem Cells International Stem Cells and Ion Channels Guest Editors: Stefan Liebau, Alexander Kleger, Michael Levin, and Shan Ping Yu Copyright © 2013 Hindawi Publishing Corporation. All rights reserved. This is a special issue published in “Stem Cells International.” All articles are open access articles distributed under the Creative Com- mons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Editorial Board Nadire N. Ali, UK Joseph Itskovitz-Eldor, Israel Pranela Rameshwar, USA Anthony Atala, USA Pavla Jendelova, Czech Republic Hannele T. Ruohola-Baker, USA Nissim Benvenisty, Israel Arne Jensen, Germany D. S. Sakaguchi, USA Kenneth Boheler, USA Sue Kimber, UK Paul R. Sanberg, USA Dominique Bonnet, UK Mark D. Kirk, USA Paul T. Sharpe, UK B. Bunnell, USA Gary E. Lyons, USA Ashok Shetty, USA Kevin D. Bunting, USA Athanasios Mantalaris, UK Igor Slukvin, USA Richard K. Burt, USA Pilar Martin-Duque, Spain Ann Steele, USA Gerald A. Colvin, USA EvaMezey,USA Alexander Storch, Germany Stephen Dalton, USA Karim Nayernia, UK Marc Turner, UK Leonard M. Eisenberg, USA K. Sue O’Shea, USA Su-Chun Zhang, USA Marina Emborg, USA J. Parent, USA Weian Zhao, USA Josef Fulka, Czech Republic Bruno Peault, USA Joel C. Glover, Norway Stefan Przyborski, UK Contents Stem Cells and Ion Channels, Stefan Liebau, -

Expression of GABAA Α2-, Β1- And
OPEN Citation: Transl Psychiatry (2013) 3, e303; doi:10.1038/tp.2013.64 & 2013 Macmillan Publishers Limited All rights reserved 2158-3188/13 www.nature.com/tp ORIGINAL ARTICLE Expression of GABAA a2-, b1- and e-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder SH Fatemi1,2,3, TD Folsom1, RJ Rooney4 and PD Thuras5 There is abundant evidence that dysfunction of the g-aminobutyric acid (GABA)ergic signaling system is implicated in the pathology of schizophrenia and mood disorders. Less is known about the alterations in protein expression of GABA receptor subunits in brains of subjects with schizophrenia and mood disorders. We have previously demonstrated reduced expression of GABAB receptor subunits 1 and 2 (GABBR1 and GABBR2) in the lateral cerebella of subjects with schizophrenia, bipolar disorder and major depressive disorder. In the current study, we have expanded these studies to examine the mRNA and protein expression of 12 GABAA subunit proteins (a1, a2, a3, a5, a6, b1, b2, b3, d, e, g2 and g3) in the lateral cerebella from the same set of subjects with schizophrenia (N ¼ 9–15), bipolar disorder (N ¼ 10–15) and major depression (N ¼ 12–15) versus healthy controls (N ¼ 10–15). We found significant group effects for protein levels of the a2-, b1- and e-subunits across treatment groups. We also found a significant group effect for mRNA levels of the a1-subunit across treatment groups. New avenues for treatment, such as the use of neurosteroids to promote GABA modulation, could potentially ameliorate GABAergic dysfunction in these disorders. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -
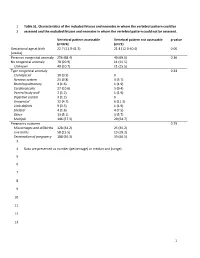
1 Table S1. Characteristics of the Included Fetuses and Neonates in Whom the Vertebral Pattern Could Be 1 Assessed and the Exclu
1 Table S1. Characteristics of the included fetuses and neonates in whom the vertebral pattern could be 2 assessed and the excluded fetuses and neonates in whom the vertebral pattern could not be assessed. Vertebral pattern assessable Vertebral pattern not assessable p-value (n=374) (n=71) Gestational age at birth 22.7 (11.9-41.3) 21.4 (12.0-40.4) 0.06 (weeks) Presence congenital anomaly 256 (68.4) 49 (69.0) 0.36 No congenital anomaly 78 (20.9) 11 (15.5) Unknown 40 (10.7) 11 (15.5) Type congenital anomaly 0.24 Craniofacial 10 (3.9) 0 Nervous system 25 (9.8) 3 (5.7) Bronchopulmonary 4 (1.6) 1 (1.9) Cardiovascular 27 (10.6) 5 (9.4) Ventral body wall 3 (1.2) 1 (1.9) Digestive system 3 (1.2) 0 Urogenital 12 (4.7) 6 (11.3) Limb defects 9 (3.5) 1 (1.9) Skeletal 4 (1.6) 4 (7.5) Other 13 (5.1) 3 (5.7) Multiple 146 (57.3) 29 (54.7) Pregnancy outcome 0.79 Miscarriages and stillbirths 128 (34.2) 25 (35.2) Live births 58 (15.5) 13 (18.3) Termination of pregnancy 188 (50.3) 33 (46.5) 3 4 Data are presented as number (percentage) or median and (range). 5 6 7 8 9 10 11 12 13 1 14 Table S2. Gestational age at birth, age at death, presence of congenital abnormalities and cause of death of the included live births. GA at birth Age at death Cervical Congenital abnormalities Cause of death (weeks) (days) rib(s) 22.3 0 No. -

The Role of Δ Subunit-Containing Γ-Aminobutyric Acid Type a Receptors
THE ROLE OF δ SUBUNIT-CONTAINING γ-AMINOBUTYRIC ACID TYPE A RECEPTORS IN MEMORY AND SYNAPTIC PLASTICITY by Paul David Whissell A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by Paul Whissell, 2014 Paul Whissell The role of δ subunit-containing γ-aminobutyric acid type A receptors in memory and synaptic plasticity Doctor of Philosophy, Institute of Medical Science, University of Toronto, 2014 Abstract Background: Extrasynaptic γ-aminobutyric acid type A receptors that contain the δ subunit (δGABAA receptors) are highly expressed in the dentate gyrus (DG) of the hippocampus, where they generate a tonic conductance that regulates activity. GABAA receptor-dependent signaling regulates memory and neurogenesis in the adult DG; however, the role of δGABAA receptors in these processes is unclear. Accordingly, it was postulated that δGABAA receptors regulate memory and neurogenesis in the DG. Methods: A combination of genetic and pharmacologic techniques was employed. Memory in wild-type (WT) and δ subunit null (Gabrd–/–) mice was assessed using object-place recognition, novel object recognition, contextual discrimination, fear conditioning, fear extinction and water maze tasks. Long-term potentiation, a molecular correlate of memory, was examined using the in vitro hippocampal slice preparation. To ascertain the effects of enhanced δGABAA receptor activity, the receptor-preferring agonist 4,5,6,7- tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP; 4 mg/kg) was applied either as a pre-treatment (2 weeks prior to testing) or an acute treatment (30 min prior to testing). Results: Gabrd–/– mice exhibited impaired object-place recognition, novel object recognition and contextual discrimination relative to WT mice.