PB #ISAG2017 1 @Isagofficial #ISAG2017 #ISAG2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Each Wild Idea: Writing, Photography, History
e “Unruly, energetic, unmastered. Also erudite, engaged and rigorous. Batchen’s essays have arrived at exactly the e a c h w i l d i d e a right moment, when we need their skepticism and imagination to clarify the blurry visual thinking of our con- a writing photography history temporary cultures.” geoffrey batchen c —Ross Gibson, Creative Director, Australian Centre for the Moving Image h In Each Wild Idea, Geoffrey Batchen explores widely ranging “In this remarkable book, Geoffrey Batchen picks up some of the threads of modernity entangled and ruptured aspects of photography, from the timing of photography’s by the impact of digitization and weaves a compelling new tapestry. Blending conceptual originality, critical invention to the various implications of cyberculture. Along w insight and historical rigor, these essays demand the attention of all those concerned with photography in par- the way, he reflects on contemporary art photography, the role ticular and visual culture in general.” i of the vernacular in photography’s history, and the —Nicholas Mirzoeff, Art History and Comparative Studies, SUNY Stony Brook l Australianness of Australian photography. “Geoffrey Batchen is one of the few photography critics equally adept at historical investigation and philosophi- d The essays all focus on a consideration of specific pho- cal analysis. His wide-ranging essays are always insightful and rewarding.” tographs—from a humble combination of baby photos and —Mary Warner Marien, Department of Fine Arts, Syracuse University i bronzed booties to a masterwork by Alfred Stieglitz. Although d Batchen views each photograph within the context of broader “This book includes the most important essays by Geoffrey Batchen and therefore is a must-have for every schol- social and political forces, he also engages its own distinctive ar in the fields of photographic history and theory. -

Use of Genomic Tools to Discover the Cause of Champagne Dilution Coat Color in Horses and to Map the Genetic Cause of Extreme Lordosis in American Saddlebred Horses
University of Kentucky UKnowledge Theses and Dissertations--Veterinary Science Veterinary Science 2014 USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES Deborah G. Cook University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Cook, Deborah G., "USE OF GENOMIC TOOLS TO DISCOVER THE CAUSE OF CHAMPAGNE DILUTION COAT COLOR IN HORSES AND TO MAP THE GENETIC CAUSE OF EXTREME LORDOSIS IN AMERICAN SADDLEBRED HORSES" (2014). Theses and Dissertations--Veterinary Science. 15. https://uknowledge.uky.edu/gluck_etds/15 This Doctoral Dissertation is brought to you for free and open access by the Veterinary Science at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Veterinary Science by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Snps) Located in Exon 1 of Kappa-Casein Gene (CSN3) in Martina Franca Donkey Breed
African Journal of Biotechnology Vol. 10(26), pp. 5118-5120, 13 June, 2011 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB10.2440 ISSN 1684–5315 © 2011 Academic Journals Short Communication Analysis of two single-nucleotide polymorphisms (SNPs) located in exon 1 of kappa-casein gene (CSN3) in Martina Franca donkey breed Maria Selvaggi* and Cataldo Dario Department of Animal Health and Welfare, University of Bari, strada prov. le per Casamassima Km 3 – 70010, Valenzano (Ba), Italy. Accepted 18 January, 2011 The aim of this study is to assess genetic polymorphism at two loci in the exon 1 of the kappa-casein gene (CSN3) in Martina Franca donkey breed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analysis. Martina Franca donkey was derived from the Catalan donkey brought to Apulia at the time of the Spanish rule. This donkey is tall and well built and has good temperament. Both considered loci were found to be monomorphic in the considered population. At CSN3/PstI locus, all the animals were genotyped as AA since no AG and GG animals were found in the population. A similar result was found at CSN3/BseYI locus: all the donkeys were monomorphic and genotyped as AA. As a consequence, only one out of nine possible combined genotype (AAAA) was detected. Key words: Martina Franca donkey, kappa-casein gene (CSN3), gene polymorphism, polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). INTRODUCTION Kappa-casein is the protein that determines the size and (Lenasi et al., 2003). CSN3 is not evolutionarily related to the specific function of milk micelles; its cleavage by the “calcium-sensitive” casein genes, but is physically chymosin is responsible for milk coagulation (Yahyaoui et linked to this gene family, and is functionally important for al., 2003). -

Dynamic Technique Analysis of the Female Equestrian Rider
Dynamic technique analysis of the female equestrian rider Céleste A. Wilkins Thesis submitted in partial fulfilment of the requirements of the University of the West of England for the degree of Doctor of Philosophy Faculty of Health and Applied Sciences/Hartpury University June 2021 1 Abstract INTRODUCTION Advances in both measurement and analysis techniques offer the opportunity to assess the dynamics of the rider’s position in the saddle. As previous studies have used discrete measures to summarise the kinematics of the rider over the movement cycle or compared riders grouped by their experience level, many questions remain on the characteristics of the rider’s dynamic technique and the influence of dressage competition level. This thesis aimed to examine the competitive dressage rider’s pelvis, progressing from its static posture to analyses of the dynamic pelvic technique, and the rider’s coordination in a controlled environment using a riding simulator. METHODS Fifty-two nationally or internationally competitive female dressage riders volunteered and were measured by motion capture in simulated walk, trot, and canter, using a riding simulator and optical motion capture. Simulator-rider coordination was measured using the continuous relative phase (CRP) and CRP variability. The effect of rider competition level on CRP variability was assessed using statistical parametric mapping. The characteristics of simulator-pelvis harmony were analysed using principal component analysis (PCA) on the CRP. Finally, riders were grouped by their trunk and pelvis coordination strategies using a self-organising map (SOM) and k-means clustering. RESULTS There were no significant correlations between static and dynamic pelvic tilt. Simulator- pelvis coordination was significantly less variable than simulator-trunk, simulator-left foot or simulator-head in medium trot. -
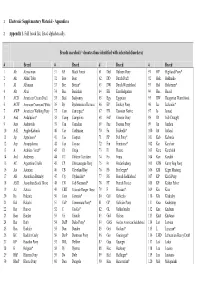
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

New Prospective on Sentinel Animal Systems: Experiences in Southern Italy Polluted Areas”
UNIVERSITÀ DEGLI STUDI DI NAPOLI “FEDERICO II” Tesi di Dottorato “New Prospective on Sentinel Animal Systems: Experiences in Southern Italy Polluted Areas” Coordinatore Candidato Tutor Prof. Giuseppe Cringoli Dott. Alessandro Costagliola Prof. Orlando Paciello To My Daughter, Ginevra. Go Forth and Pursue All Your Dreams Table of Contents List of Abbreviations 9 List of Figures 10 List of Tables 13 Abstract 15 General Introduction I Animal biomonitoring and micropollutants in public health 18 II Bioavailability and hazard of heavy metals contamination 20 II.I Lead 22 II.II Mercury 23 II.III Arsenic 25 II.IV Cadmium 27 III Environmental monitoring: bioindicators, bioaccumulators and biomarkers 29 IV Sentinel Animal Systems 32 V Selection criteria of sentinel animals in environmental biomonitoring 37 VI Domestic Animals as Sentinels 41 Bibliography 46 Chapter 1. The Role of Necropsy in Environmental Monitoring: the Regional Reference Center of Urban Veterinary Hygiene (CRIUV) experience 3 Table of Contents 1.1 Introduction 54 1.2 Materials and Methods 67 1.3 Results 74 1.4 Discussion 116 Bibliography 127 Chapter 2. Dairy cattle as a sensitive warning system for nitrate environmental pollution: evaluation of risk for human health 2.1 Introduction 135 2.2 Materials and Methods 138 2.3 Results 141 2.4 Discussion 145 Bibliography 150 Chapter 3. Animal Biomonitoring and Epidemiological Surveillance in polluted areas of Basillicata Region 3.1 Introduction 153 3.2 Materials and Methods 160 3.3 Results 164 3.4 Discussion 169 Bibliography 172 Conclusions -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Protein EVI2B Gene Symbol Evi2b Organism Mouse Gene Summary Description Not Available Gene Aliases Evi-2, Evi2 RefSeq Accession No. NC_000077.6, NT_096135.6 UniGene ID Not Available Ensembl Gene ID ENSMUSG00000093938 Entrez Gene ID 216984 Assay Information Unique Assay ID qMmuCEP0062720 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSMUST00000179322, ENSMUST00000170422 Amplicon Context Sequence ACAGTATAATCATGAGTATAGCTACCAACATAGAAATTATAATAGTACCGATTAAT ATGGCAGCAATTGCATTGTGGTTGTTTTTATGAGCAGTTCCTTTGCCACTTGGTG GTTCTGAAGTGAGTGGTGGTGGTGGTGGTGGTG Amplicon Length (bp) 114 Chromosome Location 11:79516072-79516215 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9997 cDNA Cq 22.69 cDNA Tm (Celsius) 79 gDNA Cq 24.61 Specificity (%) 49.93 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Evi2b, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

ATP Challenger Tour by the Numbers
ATP MEDIA INFORMATION Updated: 20 September 2021 2021 ATP CHALLENGER BY THE NUMBERS Match Wins Leaders W-L Titles 1) Benjamin Bonzi FRA 49-11 6 2) Tomas Martin Etcheverry ARG 38-13 2 3) Zdenek Kolar CZE 29-18 3 4) Holger Rune DEN 28-7 3 5) Kacper Zuk POL 26-11 1 Nicolas Jarry CHI 26-12 1 7) Sebastian Baez ARG 25-5 3 Altug Celikbilek TUR 25-10 2 Juan Manuel Cerundolo ARG 25-10 3 Tomas Barrios Vera CHI 25-11 1 11) Jenson Brooksby USA 23-3 3 Gastao Elias POR 23-12 1 Win Percentage Leaders W-L Pct. Titles 1) Jenson Brooksby USA 23-3 88.5 3 2) Sebastian Baez ARG 25-5 83.3 3 3) Benjamin Bonzi FRA 49-11 81.7 6 4) Holger Rune DEN 28-7 80.0 3 5) Zizou Bergs BEL 19-6 76.0 3 6) Federico Coria ARG 18-6 75.0 1 7) Tomas Martin Etcheverry ARG 38-13 74.5 2 8) Arthur Rinderknech FRA 18-7 72.0 1 Botic van de Zandschulp NED 18-7 72.0 0 *Minimum 20 matches played* Singles Title Leaders ----- By Surface ----- Player Total Clay Grass Hard Carpet Benjamin Bonzi FRA 6 1 5 Sebastian Baez ARG 3 3 Zizou Bergs BEL 3 1 2 Jenson Brooksby USA 3 1 2 Juan Manuel Cerundolo ARG 3 3 Tallon Griekspoor NED 3 3 Zdenek Kolar CZE 3 3 Holger Rune DEN 3 3 Franco Agamenone ITA 2 2 Daniel Altmaier GER 2 2 Altug Celikbilek TUR 2 2 Mitchell Krueger USA 2 2 Tomas Martin Etcheverry ARG 2 2 Mats Moraing GER 2 2 Carlos Taberner ESP 2 2 Bernabe Zapata Miralles ESP 2 2 53 tied with 1 title each Winners by Age: 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 0 0 6 7 8 4 7 10 13 13 4 3 7 5 3 1 0 3 0 1 0 1 0 0 Youngest Final: Juan Manuel Cerundolo (19) d. -

CALIFORNIA STATE UNIVERSITY, NORTHRIDGE Bioinformatic
CALIFORNIA STATE UNIVERSITY, NORTHRIDGE Bioinformatic Comparison of the EVI2A Promoter and Coding Regions A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Biology By Max Weinstein May 2020 The thesis of Max Weinstein is approved: Professor Rheem D. Medh Date Professor Virginia Oberholzer Vandergon Date Professor Cindy Malone, Chair Date California State University Northridge ii Table of Contents Signature Page ii List of Figures v Abstract vi Introduction 1 Ecotropic Viral Integration Site 2A, a Gene within a Gene 7 Materials and Methods 11 PCR and Cloning of Recombinant Plasmid 11 Transformation and Cell Culture 13 Generation of Deletion Constructs 15 Transfection and Luciferase Assay 16 Identification of Transcription Factor Binding Sites 17 Determination of Region for Analysis 17 Multiple Sequence Alignment (MSA) 17 Model Testing 18 Tree Construction 18 Promoter and CDS Conserved Motif Search 19 Results and Discussion 20 Choice of Species for Analysis 20 Mapping of Potential Transcription Factor Binding Sites 20 Confirmation of Plasmid Generation through Gel Electrophoresis 21 Analysis of Deletion Constructs by Transient Transfection 22 EVI2A Coding DNA Sequence Phylogenetics 23 EVI2A Promoter Phylogenetics 23 EVI2A Conserved Leucine Zipper 25 iii EVI2A Conserved Casein kinase II phosphorylation site 26 EVI2A Conserved Sox-5 Binding Site 26 EVI2A Conserved HLF Binding Site 27 EVI2A Conserved cREL Binding Site 28 EVI2A Conserved CREB Binding Site 28 Summary 29 Appendix: Figures 30 Literature Cited 44 iv List of Figures Figure 1. EVI2A is nested within the gene NF1 30 Figure 2 Putative Transcriptions Start Site 31 Figure 3. Gel Electrophoresis of pGC Blue cloned, Restriction Digested Plasmid 32 Figure 4. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
BCL6 maintains thermogenic capacity of brown adipose tissue during dormancy by Vassily Kutyavin DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Eric Verdin Chair ______________________________________________________________________________Ajay Chawla ______________________________________________________________________________Ethan Weiss ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2019 by Vassily Kutyavin ii Dedicated to everyone who has supported me during my scientific education iii Acknowledgements I'm very grateful to my thesis adviser, Ajay Chawla, for his mentorship and support during my dissertation work over the past five years. Throughout my time in his lab, I was always able to rely on his guidance, and his enthusiasm for science was a great source of motivation. Even when he was traveling, he could easily be reached for advice by phone or e- mail. I am particularly grateful for his help with writing the manuscript, which was probably the most challenging aspect of graduate school for me. I am also very grateful to him for helping me find a postdoctoral fellowship position. Ajay's inquisitive and fearless approach to science have been a great inspiration to me. In contrast to the majority of scientists who focus narrowly on a specific topic, Ajay pursued fundamental questions across a broad range of topics and was able to make tremendous contributions. My experience in his lab instilled in me a deep appreciation for thinking about the entire organism from an evolutionary perspective and focusing on the key questions that escape the attention of the larger scientific community. As I move forward in my scientific career, there is no doubt that I will rely on him as a role model.