Surface Freezing in N-Alkanes: Experimental and Molecular Dynamics Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je3a29 USER: ckt69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i03/DIV_je030236t DATE: March 15, 2004 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121, USA 5 6 The temperature dependence of gas-chromatographic retention times for henitriacontane to octatriacontane 7 is reported. These data are used in combination with other literature values to evaluate the vaporization 8 enthalpies and vapor pressures of these n-alkanes from T ) 298.15 to 575 K. The vapor pressure and 9 vaporization enthalpy results obtained are compared with existing literature data where possible and 10 found to be internally consistent. The vaporization enthalpies and vapor pressures obtained for the 11 n-alkanes are also tested directly and through the use of thermochemical cycles using literature values 12 for several polyaromatic hydrocarbons. Sublimation enthalpies are also calculated for hentriacontane to 13 octatriacontane by combining vaporization enthalpies with temperature adjusted fusion enthalpies. 14 15 Introduction vaporization enthalpies of C21 to C30 to include C31 to C38. 54 The vapor pressures and vaporization enthalpies of these 55 16 Polycyclic aromatic hydrocarbons (PAHs) are important larger n-alkanes have been evaluated using the technique 56 17 environmental contaminants, many of which exhibit very of correlation gas chromatography. This technique relies 57 18 low vapor pressures. In the gas phase, they exist mainly entirely on the use of standards in assessment. Standards 58 19 sorbed to aerosol particles. -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
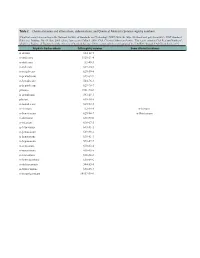
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Accuracy and Methodologic Challenges of Volatile Organic Compound–Based Exhaled Breath Tests for Cancer Diagnosis: a Systematic Review and Pooled Analysis
1 Supplementary Online Content Hanna GB, Boshier PR, Markar SR, Romano A. Accuracy and Methodologic Challenges of Volatile Organic Compound–Based Exhaled Breath Tests for Cancer Diagnosis: A Systematic Review and Pooled Analysis. JAMA Oncol. Published online August 16, 2018. doi:10.1001/jamaoncol.2018.2815 Supplement. eTable 1. Search strategy for cancer systematic review eTable 2. Modification of QUADAS-2 assessment tools eTable 3. QUADAS-2 results eTable 6. STARD assessment of each study eTable 7. Summary of factors reported to influence levels of volatile organic compounds within exhaled breath eFigure 1. Risk of bias and applicability concerns using QUADAS-2 eFigure 2. PRISMA flowchart of literature search eTable 4. Details of studies on exhaled volatile organic compounds in cancer eTable 5. Cancer VOCs in exhaled breath and their chemical class. eFigure 3. Chemical classes of VOCs reported in different tumor sites. This supplementary material has been provided by the authors to give readers additional information about their work. © 2018 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 2 eTable 1. Search strategy for cancer systematic review # Search 1 (cancer or neoplasm* or malignancy).ab. 2 limit 1 to abstracts 3 limit 2 to cochrane library [Limit not valid in Ovid MEDLINE(R),Ovid MEDLINE(R) Daily Update,Ovid MEDLINE(R) In-Process,Ovid MEDLINE(R) Publisher; records were retained] 4 limit 3 to english language 5 limit 4 to human 6 limit 5 to yr="2000 -Current" 7 limit 6 to humans 8 (cancer or neoplasm* or malignancy).ti. 9 limit 8 to abstracts 10 limit 9 to cochrane library [Limit not valid in Ovid MEDLINE(R),Ovid MEDLINE(R) Daily Update,Ovid MEDLINE(R) In-Process,Ovid MEDLINE(R) Publisher; records were retained] 11 limit 10 to english language 12 limit 11 to human 13 limit 12 to yr="2000 -Current" 14 limit 13 to humans 15 7 or 14 16 (volatile organic compound* or VOC* or Breath or Exhaled).ab. -

Phase Equilibrium Behavior of the Binary Systems CO2 + Nonadecane and CO2 + Soysolv and the Ternary System CO2 + Soysolv + Quaternary Ammonium Chloride Surfactant
J. Chem. Eng. Data 2003, 48, 1401-1406 1401 Phase Equilibrium Behavior of the Binary Systems CO2 + Nonadecane and CO2 + Soysolv and the Ternary System CO2 + Soysolv + Quaternary Ammonium Chloride Surfactant Mohamed M. Elbaccouch,† Valeriy I. Bondar,‡ Ruben G. Carbonell,* and Christine S. Grant Box 7905sChE Department, North Carolina State University, Raleigh, North Carolina 27695-7905 Liquid phase and molar volume data were measured for the binary system CO2 + soysolv at (298.15, 313.15, 323.15, 333.15, and 343.15) K and the ternary system CO2 + soysolv + quaternary ammonium chloride surfactant at (298.15, 313.15, and 333.15) K, where the composition of soysolv to the surfactant is 99:1 wt % and 80:20 wt % on a CO2-free basis. Data were collected stoichiometrically with a high- pressure Pyrex glass cell, where no sampling or chromatographic equipment is required. The accuracy of the experimental apparatus was tested with phase equilibrium measurements for the system CO2 + nonadecane at 313.15 K. A pressure-decay technique was used to calculate the mass of CO2 loaded into the equilibrium section of the apparatus, and its accuracy was verified with a blank nitrogen experiment. The generated data show that CO2 modified soysolv is an effective transport medium for the quaternary ammonium chloride surfactant. Introduction Understanding the effects of gas solubility and swollen volume on the phase behavior of a mixture is important in various thermodynamic and industrial applications. The feasibility of using CO2, modified with a food grade oil cosolvent, as an extraction medium for particular com- pounds in chemical processes is of industrial interest. -

Exhaust Emission Profiles for EPA SPECIATE Database
Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Assessment and Standards Division Office of ransportationT and Air Quality U.S. Environmental Protection Agency EPA-420-R-09-002 June 2009 Table of Contents 1.0 Introduction....................................................................................................................... 1 1.1 Background..................................................................................................................... 1 1.2 Purpose of the Project ..................................................................................................... 1 2.0 Methods.............................................................................................................................. 2 2.1 Exhaust Emissions Data.................................................................................................. 2 2.2 Calculation of Composite Profiles.................................................................................. 3 2.3 Adjustment of E0 and E10 profiles with an In-Use Gasoline......................................... 3 2.4 Assignment of SPECIATE Identification Numbers ....................................................... 4 3.0 Results ............................................................................................................................... -

UST and ASTM Petrochemical Standards
UST and ASTM Petrochemical Standards Your essential resource for Agilent ULTRA chemical standards Table of contents Introduction 3 Pennsylvania – GRO and PAH 22 About Agilent standards 3 Tennessee – GRO and DRO 22 Products 3 Texas – TNRCC Method 1005, 1006 23 Markets 3 Washington – Volatile Petroleum Hydrocarbons (VPH) method 24 Custom products 3 Washington – Extractable Petroleum Hydrocarbons Quality control laboratory 4 (EPH) method 25 Quality control validation levels 4 Washington and Oregon – Total Petroleum Hydrocarbons Triple certification 5 (NWTPH) methods 26 Level 2 reference material Certificate of Analysis 6 Maine – GRO and DRO 26 GHS compliance 7 Internal and surrogate standards for Underground Storage Tank (UST) testing 27 Underground Storage Tank (UST) Standards 8 EPA Method 1664A 28 Alaska – Method AK 101, AK 102, AK 103 10 EPA Method 418.1 28 Arizona – Method 8015AZ 11 Hydrocarbon fuel standards 29 California – PVOC and WIP 12 Weathered hydrocarbon fuel standards 30 Connecticut – ETPH method 12 EN 14105:2003 31 Florida – Method FL-PRO 13 Iowa – Methods OA-1, OA-2 14 ASTM Methods 32 Kansas – TPH method 15 ASTM Method D6584 32 Kansas modified 8015 (LRH) 15 ASTM Method D1387 33 Maine – Method 4.1.25, 4.2.17 16 ASTM Method D3710 34 Massachusetts – Volatile Petroleum Hydrocarbons ASTM Method D4815 34 (VPH) method 17 ASTM Method D5453 35/36 Massachusetts – Extractable Petroleum Hydrocarbons ASTM Methods D3120, D3246, D3961 36 (EPH) method 18 ASTM Method D4629 37 Shooters – Open and shoot spiking standards 19 ASTM Method D5762 38 Michigan – GRO and PNA 20 ASTM Method D4929 38 Mississippi – GRO, DRO, and PAH 20 ASTM Method D5808 38 New Jersey – OQA-QAM-025 21 New York – STARS compounds 22 Agilent Service and Support 39 2 UST and ASTM Petrochemical Standards Introduction About Agilent standards Agilent is a global leader in chromatography and spectroscopy, as well as an expert in chemical standards manufacturing. -

ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability Of
ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability of Coastal Plain Groundwater to Flood Water Intrusion using High Resolution Mass Spectrometry. (Under the direction of Dr. Elizabeth Guthrie Nichols). Communities in the North Carolina Coastal Plain (NCCP) depend on safe and reliable groundwater for private well use, agriculture, industry, and livelihoods. Although storm intensity and frequency are predicted to increase in coastal areas, the risk of surficial and confined aquifer contamination from extreme storms is not understood. In September 2018, Hurricane Florence caused extensive flooding across the NCCP for several weeks. The North Carolina Department of Environmental Quality (NCDEQ) Groundwater Management Branch had just completed sampling of some wells in their monitoring network when Hurricane Florence made landfall. NCDEQ returned to these wells, particularly those flooded by Hurricane Florence, for post-flood sampling. These groundwater samples were analyzed by NCDEQ for regulated semi-volatile organics with few to any detections of regulated organic contaminants. NCDEQ provided NC State the same sample extracts for analysis by high resolution mass spectrometry (HRMS). This research reports on the non-targeted and suspect-screening HRMS analyses of groundwater from nested monitoring wells. Some monitoring well sites experienced flooding during the study period, and some did not. The goal of this research was to advance our understanding of coastal aquifer susceptibility to flooding by producing the first comprehensive organic chemical profiles of coastal aquifers and by determining if aquifers have distinct organic chemical profiles that change after flooding events. This study used HRMS analyses to produce the first comprehensive organic chemical profiles of 11 aquifers in the coastal plain. -

Flammability Limits, Flash Points, and Their Consanguinity: Critical Analysis, Experimental Exploration, and Prediction
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2010-06-25 Flammability Limits, Flash Points, and Their Consanguinity: Critical Analysis, Experimental Exploration, and Prediction Jeffrey R. Rowley Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Chemical Engineering Commons BYU ScholarsArchive Citation Rowley, Jeffrey R., "Flammability Limits, Flash Points, and Their Consanguinity: Critical Analysis, Experimental Exploration, and Prediction" (2010). Theses and Dissertations. 2233. https://scholarsarchive.byu.edu/etd/2233 This Dissertation is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Flammability Limits, Flash Points, and their Consanguinity: Critical Analysis, Experimental Exploration, and Prediction Jef Rowley A dissertation submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Doctor of Philosophy W. Vincent Wilding, Chair Richard L. Rowley Larry L. Baxter Thomas A. Knotts David O. Lignell Department of Chemical Engineering Brigham Young University August 2010 Copyright © 2010 Jef Rowley All Rights Reserved ABSTRACT Flammability Limits, Flash Points, and their Consanguinity: Critical Analysis, Experimental Exploration, and Prediction Jef Rowley Department of Chemical Engineering Doctor of Philosophy Accurate flash point and flammability limit data are needed to design safe chemical processes. Unfortunately, improper data storage and reporting policies that disregard the temperature dependence of the flammability limit and the fundamental relationship between the flash point and the lower flammability limit have resulted in compilations filled with erroneous values.