C Ferrosilite and Crystal Chemistry of High-Pressure C2/ C Pyroxenes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mid-Infrared Optical Constants of Clinopyroxene and Orthoclase Derived from Oriented Single-Crystal Reflectance Spectra
American Mineralogist, Volume 99, pages 1942–1955, 2014 Mid-infrared optical constants of clinopyroxene and orthoclase derived from oriented single-crystal reflectance spectra JESSICA A. ARNOLD1,*, TIMOTHY D. GLOTCH1 AND ANNA M. PLONKA1 1Department of Geosciences, Stony Brook University, Stony Brook, New York 11794, U.S.A. ABSTRACT We have determined the mid-IR optical constants of one alkali feldspar and four pyroxene compo- sitions in the range of 250–4000 cm–1. Measured reflectance spectra of oriented single crystals were iteratively fit to modeled spectra derived from classical dispersion analysis. We present the real and imaginary indices of refraction (n and k) along with the oscillator parameters with which they were modeled. While materials of orthorhombic symmetry and higher are well covered by the current literature, optical constants have been derived for only a handful of geologically relevant monoclinic materials, including gypsum and orthoclase. Two input parameters that go into radiative transfer models, the scattering phase function and the single scattering albedo, are functions of a material’s optical constants. Pyroxene is a common rock-forming mineral group in terrestrial bodies as well as meteorites and is also detected in cosmic dust. Hence, having a set of pyroxene optical constants will provide additional details about the composition of Solar System bodies and circumstellar materials. We follow the method of Mayerhöfer et al. (2010), which is based on the Berreman 4 × 4 matrix formulation. This approach provides a consistent way to calculate the reflectance coefficients in low- symmetry cases. Additionally, while many models assume normal incidence to simplify the dispersion relations, this more general model applies to reflectance spectra collected at non-normal incidence. -

A Comparative Study of Jadeite, Omphacite and Kosmochlor Jades from Myanmar, and Suggestions for a Practical Nomenclature
Feature Article A Comparative Study of Jadeite, Omphacite and Kosmochlor Jades from Myanmar, and Suggestions for a Practical Nomenclature Leander Franz, Tay Thye Sun, Henry A. Hänni, Christian de Capitani, Theerapongs Thanasuthipitak and Wilawan Atichat Jadeitite boulders from north-central Myanmar show a wide variability in texture and mineral content. This study gives an overview of the petrography of these rocks, and classiies them into ive different types: (1) jadeitites with kosmochlor and clinoamphibole, (2) jadeitites with clinoamphibole, (3) albite-bearing jadeitites, (4) almost pure jadeitites and (5) omphacitites. Their textures indicate that some of the assemblages formed syn-tectonically while those samples with decussate textures show no indication of a tectonic overprint. Backscattered electron images and electron microprobe analyses highlight the variable mineral chemistry of the samples. Their extensive chemical and textural inhomogeneity renders a classiication by common gemmological methods rather dificult. Although a deinitive classiication of such rocks is only possible using thin-section analysis, we demonstrate that a fast and non-destructive identiication as jadeite jade, kosmochlor jade or omphacite jade is possible using Raman and infrared spectroscopy, which gave results that were in accord with the microprobe analyses. Furthermore, current classiication schemes for jadeitites are reviewed. The Journal of Gemmology, 34(3), 2014, pp. 210–229, http://dx.doi.org/10.15506/JoG.2014.34.3.210 © 2014 The Gemmological Association of Great Britain Introduction simple. Jadeite jade is usually a green massive The word jade is derived from the Spanish phrase rock consisting of jadeite (NaAlSi2O6; see Ou Yang, for piedra de ijada (Foshag, 1957) or ‘loin stone’ 1999; Ou Yang and Li, 1999; Ou Yang and Qi, from its reputed use in curing ailments of the loins 2001). -

Minerals Found in Michigan Listed by County
Michigan Minerals Listed by Mineral Name Based on MI DEQ GSD Bulletin 6 “Mineralogy of Michigan” Actinolite, Dickinson, Gogebic, Gratiot, and Anthonyite, Houghton County Marquette counties Anthophyllite, Dickinson, and Marquette counties Aegirinaugite, Marquette County Antigorite, Dickinson, and Marquette counties Aegirine, Marquette County Apatite, Baraga, Dickinson, Houghton, Iron, Albite, Dickinson, Gratiot, Houghton, Keweenaw, Kalkaska, Keweenaw, Marquette, and Monroe and Marquette counties counties Algodonite, Baraga, Houghton, Keweenaw, and Aphrosiderite, Gogebic, Iron, and Marquette Ontonagon counties counties Allanite, Gogebic, Iron, and Marquette counties Apophyllite, Houghton, and Keweenaw counties Almandite, Dickinson, Keweenaw, and Marquette Aragonite, Gogebic, Iron, Jackson, Marquette, and counties Monroe counties Alunite, Iron County Arsenopyrite, Marquette, and Menominee counties Analcite, Houghton, Keweenaw, and Ontonagon counties Atacamite, Houghton, Keweenaw, and Ontonagon counties Anatase, Gratiot, Houghton, Keweenaw, Marquette, and Ontonagon counties Augite, Dickinson, Genesee, Gratiot, Houghton, Iron, Keweenaw, Marquette, and Ontonagon counties Andalusite, Iron, and Marquette counties Awarurite, Marquette County Andesine, Keweenaw County Axinite, Gogebic, and Marquette counties Andradite, Dickinson County Azurite, Dickinson, Keweenaw, Marquette, and Anglesite, Marquette County Ontonagon counties Anhydrite, Bay, Berrien, Gratiot, Houghton, Babingtonite, Keweenaw County Isabella, Kalamazoo, Kent, Keweenaw, Macomb, Manistee, -

Diopside Camgsi2o6 C 2001 Mineral Data Publishing, Version 1.2 ° Crystal Data: Monoclinic
Diopside CaMgSi2O6 c 2001 Mineral Data Publishing, version 1.2 ° Crystal Data: Monoclinic. Point Group: 2=m: As prismatic crystals with nearly square cross sections, to 50 cm; granular, columnar, lamellar massive. Twinning: Simple or multiple twins on 100 or 010 , common. f g f g Physical Properties: Cleavage: Distinct on 110 , (110) (110) 87±; partings on 100 and probably 010 . Fracture: Uneven to conchofidal.g Tenaci^ty: Britt»le. Hardness = 5f.5{6.g5 D(meas.) = 3.f22{3g.38 D(calc.) = 3.278 Optical Properties: Transparent to opaque. Color: Colorless, white, yellow, pale to dark green, black; colorless in thin section. Streak: White, gray, gray-green. Luster: Vitreous or dull. Optical Class: Biaxial (+). Orientation: Y = b; Z c = 38± on (010); X a = 22±. ^ ¡ ^ ¡ Dispersion: r > v; weak to moderate. ® = 1.664 ¯ = 1.672 ° = 1.694 2V(meas.) = 59± Cell Data: Space Group: C2=c: a = 9.746 b = 8.899 c = 5.251 ¯ = 105:63± Z = 4 X-ray Powder Pattern: Schwartzenstein, Austria. (ICDD 11-654). 2.991 (100), 2.528 (40), 2.893 (30), 2.518 (30), 3.23 (25), 2.952 (25), 1.625 (25) Chemistry: (1) (2) (1) (2) (1) (2) SiO2 54.66 54.09 FeO 0.07 1.47 K2O 0.15 + TiO2 0.28 MnO 0.02 0.09 H2O 0.22 0.22 Al2O3 0.07 1.57 MgO 18.78 16.96 H2O¡ 0.08 Fe2O3 0.68 0.74 CaO 25.85 21.10 rem: 0.49 Cr2O3 2.03 Na2O 1.37 Total 100.35 100.64 3+ (1) Juva, Finland; corresponds to Ca1:00(Mg1:01Fe0:02)§=1:03Si1:98O6: (2) Dutoitspan mine, 2+ Kimberley, Cape Province, South Africa; corresponds to (Ca0:82Na0:05Fe0:04Mg0:04K0:01)§=0:96 3+ (Mg0:88Cr0:06Al0:03Fe0:02Ti0:01)§=1:00(Si1:96Al0:04)§=2:00O6: Polymorphism & Series: Forms two series, with hedenbergite, and with johannsenite. -

Fayalite and Clinopyroxene in the Porphyries Of
Canadion Mineralogist Yol. 22, PP. zt0l-409(1984) FAYALITEAND CLINOPYROXENEIN THE PORPHYRIESOF THE TIBCHI ANOROGENIC RING-COMPLEX,NIGERIA: POSTMAGMATIC INITIATION OF A PERALKALTNETREND ECHEFUC. IKE DE)artmentof Geologt,Ahmadu Bello University,Zoria, Nigerip PETER BOWDEN Depafimentof Geology,University of St. Andrews, St.Andrews, Fife, ScottandKy16 gST ROBERT F. MARTIN Deportmentof GeologicalSciences, McGill University,3450 tlniversity Street,Montreal, QuebecH3A 2A7 ABSTRACT enAc i unephase fluide capablede s'infiltrer le long des clivages,La durdede I'interactionavec cette phase fluide The quanz and granite porphyries of the Tibchi interstitiellea 6t€la plus longuedans le casdu porphyre anorogenicring-complex, Nigeria, and their monzonitic and granitique.Le magmaa 6t€mis enplace tranqrillement le syenitic enclaves, contain fayalite and a clinopyroxene long d'une faille annulaireapr0s 6vacuation de la partie whose composition varies from ferroaugite to fer- supdrieurede la chambremagmatique par fluidisation. rohedenbergiteto sodian hedenbergite.The fayalite, which Compardeau porphyrei ph6nocristauxde quartz, qui a is unzoned, ranges from Fa92to Faeein composition; this r6sult6de cettemise en placesoudaine, le porphyregra- spread allows the ranking of the quartz porphyry and its nitiquene contientaucune trace de fayaliteet montreune enclavesin a crystallization sequence.The Fe content of r6actionplus pouss€e entre les compositions magmatiques pyroxene grains increases outward; the most evolved depyroxEne, form€es d'abord, et la phasefluide rdsiduelle. maernatic composition crystallized as hedenbergiterather A Tibchi, le caractbrehlperalcalin s'est manifestd au stade than as B-wollastonite.The proportion of the Ac compo- post-magmatiquede cristallisation. nent, which is low during most of the crystallization inter- val, increasesmarkedly at alate stage; the formation of Mot*clls: porphyred quartz,porphyre granitique, fayalite, the rim is attributed to a residual fluid phase that could h6denbergite,acmite, hyperalcalinit6, stade penetratealong cleavageplanes. -

Miissbauer Spectra of Synthetic Hedenbergite-Ferrosilite Pyroxenes'
Amefican Min.eralogist, Volume 58, pages 850-868, 1973 MiissbauerSpectra of Synthetic Hedenbergite-ferrosilitePyroxenes' ERrc Dowry, U. S. Geological Suruey, Washington, D. C. D. H. LrNnsrEys Carnegie Institution ol Washington, llashington, D. C. Abstract The hedenbergite-ferrosiliteseries may be taken as representativein some respects of other sections across the common pyroxens quadrilateral. Data are reported for eight clinopyroxenes synthesized at a temperature above the augite-pigeonitesolvus, plus two orthopyroxenesnear ferrosilite in composition.Clinopyroxenes less calcic than about Fse,Wor were found to have the pigeonite structure (p21/c) on quenching. The assignmentof doublets and the interpretation of area ratios in the M<issbauerroom- temperature spectra is clarified with the hypothesis that a distinct doublet arises from each of the four different types of next-nearest-neighborconfigurations for iron cations in site Ml. These four types arise from random distribution of calcium and iron in M2. Overlap of some of these doublets with the M2 doublet causesthe area anomaly (overestimation of M2 area) previously noted when only two doublets are fitted. This overlap apparently also influences area ratios of liquid-nitrogen temperature spectra of high-calcium pyroxenes, but in these spectra, it does not seem to be possible to fit all the different Ml doublets, owing to their close superposition.This makes it questionablewhether very accurate site distributions for high-calcium clinopyroxenescan be obtained from Mtissbauerspectra, even at liquid-nitrogen temperature. The quadrupole splitting of both M sites can be rationalized with Ingalls' crystal-field model. The Ml site, becauseof its low distortion, is very sensitive to small changes in its environment, hence the several doublets for different next-nearest-neighborconfigurations. -

Interpreting the Origins and Evolutions of Martian Basalts from Pyroxene Composition: II
Lunar and Planetary Science XXVIII 1560.PDF Interpreting the Origins and Evolutions of Martian Basalts from Pyroxene Composition: II. Vibrational Spectroscopy of Clinopyroxenes and Terrestrial Basaltic Rocks; V. E. Hamilton and P. R. Christensen, Department of Geology, Box 871404, Arizona State University, Tempe, AZ 85287- 1404 (e-mail: [email protected]). Introduction. The Thermal Emission two structural groups (Figure 1) [10]. Furthermore, we Spectrometer (TES) on the Mars Global Surveyor find that the minerals of the clinopyroxene solid spacecraft will return thermal IR spectral data of solution series (diopside, augite, and hedenbergite) martian surface materials for the purposes of can also be distinguished on the basis of their spectral geological mapping. Our goal is to quickly and characteristics (Figure 2). In the 1200 - 800 cm-1 accurately derive the compositions of martian basalts region, all three minerals display two broad groups of from these data once spectra of the martian surface absorptions, one centered at approximately 1075 cm-1 are acquired. We are preparing to interpret these data and one around 900 cm-1. The spectra of the pure by examining the spectral characteristics of pyroxenes endmembers diopside and hedenbergite contain an (common minerals in mafic compositions) and igneous additional small yet distinctive feature at 1010 or 1000 rocks. Pyroxenes ranging in composition from augite cm-1. This trough is not apparent in the spectrum of (clinopyroxene) to low-Ca orthopyroxene have been augite, suggesting that the changes in mineral identified on the martian surface in studies of visible structure (and vibrational character) with Mg/Fe and near-infrared spectroscopic data [1-4]. -

RE-EXAMINATION of the WOLLASTONITE- HEDENBERGITE (Casios-Cafesizoo)EQUILIBRIA
THE AMERICAN MINERALOGIST, VOL 56, NOVEMBER_DECEMBER, T971 RE-EXAMINATION OF THE WOLLASTONITE- HEDENBERGITE (CaSiOs-CaFeSizOo)EQUILIBRIA MenrrN S. RursrerN, Department oJ GeologicolSciences, S.U.lf.f. Collegeat I{ew Paltz, New Paltz, l{.Y. 12561 ABSTRACT The equilibria for the iron-rich wollastonite solid solution*less iron-rich wollastonite solid solution*hedenbergite reaction has been determined in the temperature interval 600"-995.C, Pnso:1kbar, and/o, defined by the QFM bufier. The equilibria develops an inflection around 800oC for a composition of (Cao.ssl-e6.12)SiO:.The flattening continues until a composition of (Cae soFeo:o)SiOaat approximately 815'C. The slope then changes smoothly to a composition of (cao sFes)Sior at 980'C. X-ray difiraction analysis of the wollastonite solid solutions from the inflected region show deveiopment of multiple re- flections analogous to all previously occurring major reflections. The X-ray data and infrared spectra measurements (Rutstein and white, 1971) indi- cate the equilibria inflection and double reflections are due to the co-existence of a wollasto- nite structure and a bustamite structure. These structures coexist with hedenbergite as a function of bulk composition and temperature Crystal chemical considerations require that both pyroxenoid structural variants undergo a series of ordered and disordered state aS a function of composition and tempera- ture. fNrnotucrroN The major previous phaseequilibria studieson the CaSiOa-CaFeSizOo join were conductedby Bowen, Schairer,and Posjnak (1933) as part of their pioneering study of the CaO-FeO-SiOzsystem. Their dry synthesis weremade at an atmosphericpressure and at an oxygen partial pressure approximating that of the fayalite-quartz-iron buffer. -

High-Temperature Crystal Chemistry of Acmite, Diopside, Hedenbergite, Jadeite, Spodumene, and Ureyite
American Mineralogist, Volume 58, pages 594-618, 1973 High-Temperature Crystal Chemistry of Acmite, Diopside, Hedenbergite, Jadeite, Spodumene, and Ureyite MARYELLEN CAMERON, SHIGEHO SUENO, C. T. PREWITT, AND J. J. PAPIKE Department of Earth and Space Sciences, State University of New York, Stony Brook, New York 11790 Abstract Crystal structure parameters have been determined for acmite, NaFe'+Si20. (at _400°C, 600°C, 800°C), diopside, CaMgSLO. (at _400°C, 700°C, 850°C, lOOO°C), hedenbergite, CaFe'+Si20. (at _24°C, 400°C, 600°C, 800°C, 900°C, lOOO°C), jadeite, NaAISi20. (at _24°C, 400°C, 600°C, 800°C), spodumene, LiAISLO. (at _300°C, 460°C, 760°C) and ureyite, NaCrSi,O. (at _400°C, 600°C). Refinements in space group C2/c using anisotropic tempera- ture factors and over 640 reflections for each set of data resulted in unweighted R-factors ranging from 0.022 to 0.078. The increase in mean Si-O bond lengths with increasing temperature is not statistically sig- nificant, but the mean M-O distances for both the 6- and 8-coordinated sites in the six pyroxenes exhibit relatively large increases. The mean thermal expansion coefficients for the Fe2+-O various bonds increase in the following order: Si'+-O < Cr3+-O < Fe"+-O < AP+-O < < Na+-O < Mg'+-O < Ca2+-O < Li+-O. This series is essentially a function of decreasing bond strength, although the position of Mg2+_O may indicate an exception to this trend. The pyroxene structures accommodate the differential mean M-O expansions through (1) exten- sion of the silicate tetrahedral chains, (2) distortion of the silicate tetrahedra, and (3) an increase in out-of-plane tilting of silicate tetrahedra. -
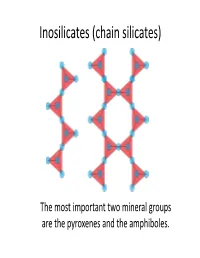
Inosilicates (Chain Silicates)
Inosilicates (chain silicates) The most important two mineral groups are the pyroxenes and the amphiboles. Pyroxenes Pyroxene General Formula (c ha in s ilicat es) XYT2O6 or M1M2(SiAl)2O6 Orthopyroxenes Clinopyroxenes (Orthorhombic) (Monoclinic) X(M2) = Na, Ca, Mn Fe, Mg and Li Y(M1) = Mn, Fe, (FeMg) Si O Ca(MgFe)Si O Al, Cr and Ti. 2 2 6 2 6 T = Si and Al. •The most important group of rock‐ forming ferromagnesian silicate Ca Si O minerals. They occur in almost every 2 2 6 type of igneous rock. Orthopyroxenes Ca2Si2O6 Magnesium‐iron The M1 and M2 sites are pyroxenes in which other essentially somewhat cations occupy less than distorted octahedrally 10% of the M1‐M2 sites. coordinated cations. (M1 is more regular than M2) 5 mol % 5 mol % Enstatite Ferrosilite 50 mol % Mg2Si2O6 Fe2Si2O6 Clinopyroxenes Wollastonite Ca2Si2O6 Magnesium‐iron pyroxenes in which Ca cations occupy The grey areas 50% of octahedral sites. represent miscbility ggpaps. (All of the M2 sites). 50 mo l % Ca Mg Si2O6 Diopside Hedenbergite CFSiCaFeSi2O6 Augite 55 mol % 10 mol % Pigeonite 5 mol % 50 mol % Mg2Si2O6 Fe2Si2O6 In special circumstances a complex coupled substitution involving both of the M‐sites will occur. Aegirine 3+ NaFe [Si2O6] Pressure increase Jaedeite AitAugite Omphacite (Na,Al)[Si2O6] (Ca,Mg,Fe,Al)[Si2O6] (Ca,Na)(Mg,Fe,Al)[Si2O6] Structure of the pyroxenes Pyroxene group minerals are made up of silicon‐oxygen tetrahedral linked at corners to form infinite chains parallel to the c axis. The chains are laterally linked by the 6‐coordinated polyhedral M‐sites. -

Minerals of Arizona Report
MINERALS OF ARIZONA by Frederic W. Galbraith and Daniel J. Brennan THE ARIZONA BUREAU OF MINES Price One Dollar Free to Residents of Arizona Bulletin 181 1970 THE UNIVERSITY OF ARIZONA TUCSON TABLE OF CONT'ENTS EIements .___ 1 FOREWORD Sulfides ._______________________ 9 As a service about mineral matters in Arizona, the Arizona Bureau Sulfosalts ._. .___ __ 22 of Mines, University of Arizona, is pleased to reprint the long-standing booklet on MINERALS OF ARIZONA. This basic journal was issued originally in 1941, under the authorship of Dr. Frederic W. Galbraith, as Simple Oxides .. 26 a bulletin of the Arizona Bureau of Mines. It has moved through several editions and, in some later printings, it was authored jointly by Dr. Gal Oxides Containing Uranium, Thorium, Zirconium .. .... 34 braith and Dr. Daniel J. Brennan. It now is being released in its Fourth Edition as Bulletin 181, Arizona Bureau of Mines. Hydroxides .. .. 35 The comprehensive coverage of mineral information contained in the bulletin should serve to give notable and continuing benefits to laymen as well as to professional scientists of Arizona. Multiple Oxides 37 J. D. Forrester, Director Arizona Bureau of Mines Multiple Oxides Containing Columbium, February 2, 1970 Tantaum, Titanium .. .. .. 40 Halides .. .. __ ____ _________ __ __ 41 Carbonates, Nitrates, Borates .. .... .. 45 Sulfates, Chromates, Tellurites .. .. .. __ .._.. __ 57 Phosphates, Arsenates, Vanadates, Antimonates .._ 68 First Edition (Bulletin 149) July 1, 1941 Vanadium Oxysalts ...... .......... 76 Second Edition, Revised (Bulletin 153) April, 1947 Third Edition, Revised 1959; Second Printing 1966 Fourth Edition (Bulletin 181) February, 1970 Tungstates, Molybdates.. _. .. .. .. 79 Silicates ... -

Significant Lunar Minerals Mineral Names the Compositions of Minerals Naturally Fall Into Certain Groups
Significant Lunar Minerals Mineral Names The compositions of minerals naturally fall into certain groups. A group shares a common anion and similarities in how the lattice of the minerals are structured. Plagioclase, olivine and pyroxene groups are silicates. Other groups of importance on the Moon are the oxides, of which the spinel group is a specific subset, the sulfides, the phosphates and the native (no anions) metals. In many cases within a group individual crystals may have quite a range of compositions. So for example a specimen in the olivine group may actually be half way between pure fayalite (Fe2SiO4) and pure forsterite (Mg2SiO4), or anywhere in between the end members. For mineral groups where this happens, geologists frequently will refer to the group name, rather than the mineral name. In such cases geologist will indicate the relative amount of each end member by giving the ratio of one end member to the total. For example an olivine that is half forsterite and half fayalite is described as Fo50, where Fo refers to the forsterite end member. This approach is also used for the plagioclase group, where the two end members are anorthite (CaAl2Si2O8) and albite (NaAlSi3O8). The relative amounts of these two end members in a crystal are expressed as An90. This is saying that 90% of the plagioclase is the anorthite end member. Lunar Abundance The terms indicating relative abundances in Table 1 are not formally defined. For this table the general concepts are as follows. Something that is Abundant is wide spread and usually more than ~33% of the material.