Isolation, Partial Purification and Characterization of An
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Key Enzymes Involved in the Synthesis of Hops Phytochemical Compounds: from Structure, Functions to Applications
International Journal of Molecular Sciences Review Key Enzymes Involved in the Synthesis of Hops Phytochemical Compounds: From Structure, Functions to Applications Kai Hong , Limin Wang, Agbaka Johnpaul , Chenyan Lv * and Changwei Ma * College of Food Science and Nutritional Engineering, China Agricultural University, 17 Qinghua Donglu Road, Haidian District, Beijing 100083, China; [email protected] (K.H.); [email protected] (L.W.); [email protected] (A.J.) * Correspondence: [email protected] (C.L.); [email protected] (C.M.); Tel./Fax: +86-10-62737643 (C.M.) Abstract: Humulus lupulus L. is an essential source of aroma compounds, hop bitter acids, and xanthohumol derivatives mainly exploited as flavourings in beer brewing and with demonstrated potential for the treatment of certain diseases. To acquire a comprehensive understanding of the biosynthesis of these compounds, the primary enzymes involved in the three major pathways of hops’ phytochemical composition are herein critically summarized. Hops’ phytochemical components impart bitterness, aroma, and antioxidant activity to beers. The biosynthesis pathways have been extensively studied and enzymes play essential roles in the processes. Here, we introduced the enzymes involved in the biosynthesis of hop bitter acids, monoterpenes and xanthohumol deriva- tives, including the branched-chain aminotransferase (BCAT), branched-chain keto-acid dehydroge- nase (BCKDH), carboxyl CoA ligase (CCL), valerophenone synthase (VPS), prenyltransferase (PT), 1-deoxyxylulose-5-phosphate synthase (DXS), 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR), Geranyl diphosphate synthase (GPPS), monoterpene synthase enzymes (MTS), cinnamate Citation: Hong, K.; Wang, L.; 4-hydroxylase (C4H), chalcone synthase (CHS_H1), chalcone isomerase (CHI)-like proteins (CHIL), Johnpaul, A.; Lv, C.; Ma, C. -

ATP-Citrate Lyase Has an Essential Role in Cytosolic Acetyl-Coa Production in Arabidopsis Beth Leann Fatland Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2002 ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis Beth LeAnn Fatland Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Molecular Biology Commons, and the Plant Sciences Commons Recommended Citation Fatland, Beth LeAnn, "ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis " (2002). Retrospective Theses and Dissertations. 1218. https://lib.dr.iastate.edu/rtd/1218 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis by Beth LeAnn Fatland A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Plant Physiology Program of Study Committee: Eve Syrkin Wurtele (Major Professor) James Colbert Harry Homer Basil Nikolau Martin Spalding Iowa State University Ames, Iowa 2002 UMI Number: 3158393 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Synthesis of Unnatural Alkaloid Scaffolds by Exploiting Plant Polyketide Synthase
Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase Hiroyuki Moritaa,b, Makoto Yamashitaa, She-Po Shia,1, Toshiyuki Wakimotoa,b, Shin Kondoc, Ryohei Katoc, Shigetoshi Sugioc,2, Toshiyuki Kohnod,2, and Ikuro Abea,b,2 aGraduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan; bJapan Science and Technology Agency, Core Research of Evolutional Science and Technology, 5 Sanbancho, Chiyoda-ku, Tokyo 102-0075, Japan; cBiotechnology Laboratory, Mitsubishi Chemical Group Science and Technology Research Center Inc., 1000 Kamoshida, Aoba, Yokohama, Kanagawa 227-8502, Japan; and dMitsubishi Kagaku Institute of Life Sciences, 11 Minamiooya, Machida, Tokyo 194-8511, Japan Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved July 12, 2011 (received for review May 15, 2011) HsPKS1 from Huperzia serrata is a type III polyketide synthase (PKS) club moss Huperzia serrata (HsPKS1) (19) (Fig. S1), using precur- with remarkable substrate tolerance and catalytic potential. Here sor-directed and structure-based approaches. As previously re- we present the synthesis of unnatural unique polyketide–alkaloid ported, HsPKS1 is a unique type III PKS that exhibits unusually hybrid molecules by exploiting the enzyme reaction using precur- broad substrate specificity to produce various aromatic polyke- sor-directed and structure-based approaches. HsPKS1 produced tides. For example, HsPKS1, which normally catalyzes the sequen- novel pyridoisoindole (or benzopyridoisoindole) with the 6.5.6- tial condensations of 4-coumaroyl-CoA (1) with three molecules fused (or 6.6.5.6-fused) ring system by the condensation of 2-carba- of malonyl-CoA (2) to produce naringenin chalcone (3)(Fig.1A), moylbenzoyl-CoA (or 3-carbamoyl-2-naphthoyl-CoA), a synthetic also accepts bulky N-methylanthraniloyl-CoA (4)asastarterto nitrogen-containing nonphysiological starter substrate, with two produce 1,3-dihydroxy-N-methylacridone (5), after three conden- molecules of malonyl-CoA. -

Two Chalcone Synthase Isozymes Participate Redundantly in UV-Induced Sakuranetin Synthesis in Rice
International Journal of Molecular Sciences Article Two Chalcone Synthase Isozymes Participate Redundantly in UV-Induced Sakuranetin Synthesis in Rice 1, 1 1 2 1, Hye Lin Park y, Youngchul Yoo , Seong Hee Bhoo , Tae-Hoon Lee , Sang-Won Lee * and Man-Ho Cho 1,* 1 Graduate School of Biotechnology and Department of Genetic Engineering, Kyung Hee University, Yongin 17104, Korea; [email protected] (H.-L.P.); [email protected] (Y.Y.); [email protected] (S.H.B.) 2 Department of Applied Chemistry, Kyung Hee University, Yongin 17104, Korea; [email protected] * Correspondence: [email protected] (S.-W.L.); [email protected] (M.-H.C.) Present address; Department of Botany and Plant Pathology, and Center for Plant Biology, y Purdue University, West Lafayette, IN 47907, USA Received: 28 April 2020; Accepted: 25 May 2020; Published: 27 May 2020 Abstract: Chalcone synthase (CHS) is a key enzyme in the flavonoid pathway, participating in the production of phenolic phytoalexins. The rice genome contains 31 CHS family genes (OsCHSs). The molecular characterization of OsCHSs suggests that OsCHS8 and OsCHS24 belong in the bona fide CHSs, while the other members are categorized in the non-CHS group of type III polyketide synthases (PKSs). Biochemical analyses of recombinant OsCHSs also showed that OsCHS24 and OsCHS8 catalyze the formation of naringenin chalcone from p-coumaroyl-CoA and malonyl-CoA, while the other OsCHSs had no detectable CHS activity. OsCHS24 is kinetically more efficient than OsCHS8. Of the OsCHSs, OsCHS24 also showed the highest expression levels in different tissues and developmental stages, suggesting that it is the major CHS isoform in rice. -

Transporters in Plant Sulfur Metabolism
REVIEW ARTICLE published: 09 September 2014 doi: 10.3389/fpls.2014.00442 Transporters in plant sulfur metabolism Tamara Gigolashvili 1* and Stanislav Kopriva 2 1 Department of Plant Molecular Physiology, Botanical Institute and Cluster of Excellence on Plant Sciences, Cologne Biocenter, University of Cologne, Cologne, Germany 2 Plant Biochemistry Department, Botanical Institute and Cluster of Excellence on Plant Sciences, Cologne Biocenter, University of Cologne, Cologne, Germany Edited by: Sulfur is an essential nutrient, necessary for synthesis of many metabolites. The uptake of Nicole Linka, Heinrich-Heine sulfate, primary and secondary assimilation, the biosynthesis, storage, and final utilization Universität Düsseldorf, Germany of sulfur (S) containing compounds requires a lot of movement between organs, cells, Reviewed by: and organelles. Efficient transport systems of S-containing compounds across the internal Viktor Zarsky, Charles University, Czech Republic barriers or the plasma membrane and organellar membranes are therefore required. Here, Frédéric Marsolais, Agriculture and we review a current state of knowledge of the transport of a range of S-containing Agri-Food Canada, Canada metabolites within and between the cells as well as of their long distance transport. An *Correspondence: improved understanding of mechanisms and regulation of transport will facilitate successful Tamara Gigolashvili, Department of engineering of the respective pathways, to improve the plant yield, biotic interaction and Plant Molecular Physiology, -

Promoter Analysis of the Chalcone Synthase ( <Emphasis Type="Italic
Plant Molecular Biology 15: 95-109, 1990. © 1990 KluwerAcademic Publishers. Printed in Belgium. 95 Promoter analysis of the chalcone synthase (chsA) gene of Petunia hybrida: a 67 bp promoter region directs flower-specific expression Ingrid M. van der Meer, Cornelis E. Spelt, Joseph N. M. Mol and Antoine R. Stuitje Department of Genetics, Vrije Universiteit, de Boelelaan 1087, 1081 HV Amsterdam, Netherlands Received 8 December 1989; accepted in revised form 3 April 1990 Key words: chalcone synthase gene (A), flower-specific transient expression, Petunia hybrida, promoter analysis, TACPyAT sequence Abstract In order to scan the 5' flanking region of the chalcone synthase (chs A) gene for regulatory sequences involved in directing flower-specific and UV-inducible expression, a chimaeric gene was constructed containing the chs A promoter of Petunia hybrida (V30), the chloramphenicol acetyl transferase (cat) structural sequence as a reporter gene and the chs A terminator region of Petunia hybrida (V30). This chimaeric gene and 5' end deletions thereof were introduced into Petunia plants with the help of Ti plasmid-derived plant vectors and CAT activity was measured. A 220 bp chs A promoter fragment contains cis-acting elements conferring flower-specific and UV-inducible expression. A promoter frag- ment from - 67 to + 1, although at a low level, was still able to direct flower-specific expression but could not drive UV-inducible expression in transgenic Petunia seedlings. Molecular analysis of binding of flower nuclear proteins to chs A promoter fragments by gel retardation assays showed strong specific binding to the sequences from - 142 to + 81. Promoter sequence comparison of chs genes from other plant species, combined with the deletion analysis and gel retardation assays, strongly suggests the involvement of the TACPyAT repeats ( - 59 and - 52) in the regulation of organ-specificity of the chs A gene in Petunia hybrida. -

Compatible and Incompatible Pollen-Styles Interaction in Pyrus Communis L
fpls-10-00741 June 12, 2019 Time: 19:21 # 1 ORIGINAL RESEARCH published: 07 June 2019 doi: 10.3389/fpls.2019.00741 Compatible and Incompatible Pollen-Styles Interaction in Pyrus communis L. Show Different Transglutaminase Features, Polyamine Pattern and Metabolomics Profiles Manuela Mandrone1, Fabiana Antognoni2, Iris Aloisi3, Giulia Potente2, Ferruccio Poli1, Giampiero Cai4, Claudia Faleri4, Luigi Parrotta3 and Stefano Del Duca3* 1 Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy, 2 Department for Life Quality Studies, Edited by: University of Bologna, Rimini, Italy, 3 Department of Biological, Geological and Environmental Sciences, University Antonio F. Tiburcio, of Bologna, Bologna, Italy, 4 Department of Life Sciences, University of Siena, Siena, Italy University of Barcelona, Spain Reviewed by: Pollen-stigma interaction is a highly selective process, which leads to compatible Penglin Sun, University of California, Riverside, or incompatible pollination, in the latter case, affecting quantitative and qualitative United States aspects of productivity in species of agronomic interest. While the genes and the Milagros Bueno, Universidad de Jaén, Spain corresponding protein partners involved in this highly specific pollen-stigma recognition *Correspondence: have been studied, providing important insights into pollen-stigma recognition in self- Stefano Del Duca incompatible (SI), many other factors involved in the SI response are not understood [email protected] yet. This work concerns the study of transglutaminase (TGase), polyamines (PAs) Specialty section: pattern and metabolomic profiles following the pollination of Pyrus communis L. This article was submitted to pistils with compatible and SI pollen in order to deepen their possible involvement in Plant Metabolism the reproduction of plants. -
Generate Metabolic Map Poster
Authors: Zheng Zhao, Delft University of Technology Marcel A. van den Broek, Delft University of Technology S. Aljoscha Wahl, Delft University of Technology Wilbert H. Heijne, DSM Biotechnology Center Roel A. Bovenberg, DSM Biotechnology Center Joseph J. Heijnen, Delft University of Technology An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Marco A. van den Berg, DSM Biotechnology Center Peter J.T. Verheijen, Delft University of Technology Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. PchrCyc: Penicillium rubens Wisconsin 54-1255 Cellular Overview Connections between pathways are omitted for legibility. Liang Wu, DSM Biotechnology Center Walter M. van Gulik, Delft University of Technology L-quinate phosphate a sugar a sugar a sugar a sugar multidrug multidrug a dicarboxylate phosphate a proteinogenic 2+ 2+ + met met nicotinate Mg Mg a cation a cation K + L-fucose L-fucose L-quinate L-quinate L-quinate ammonium UDP ammonium ammonium H O pro met amino acid a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar a sugar K oxaloacetate L-carnitine L-carnitine L-carnitine 2 phosphate quinic acid brain-specific hypothetical hypothetical hypothetical hypothetical -

1 Plant and Cell Physiology Advance Access Published April 22, 2015
Plant and Cell Physiology Advance Access published April 22, 2015 HAG3, a Histone Acetyltransferase, affects UV-B Responses by negatively regulating the Expression of DNA Repair Enzymes and Sunscreen Content in Arabidopsis thaliana Running head: GNAT family acetyltransferases in UV-B responses Corresponding author: Dr. P. Casati, e-mail [email protected]. Centro de Estudios Fotosintéticos y Bioquímicos (CEFOBI), Universidad Nacional de Rosario, Suipacha 531, 2000 Rosario, Argentina. TE/FAX 054-341-4371955 Downloaded from Subject Area: Environmental and stress responses http://pcp.oxfordjournals.org/ Number of black and white figures: 6 Number of tables: 1 Type and number of supplementary material: 3 Supplementary Figures by guest on October 27, 2016 © The Author 2015. Published by Oxford University Press on behalf of Japanese Society of Plant Physiologists. All rights reserved. For Permissions, please e-mail: [email protected] 1 HAG3, a Histone Acetyltransferase, affects UV-B Responses by negatively regulating the Expression of DNA Repair Enzymes and Sunscreen Content in Arabidopsis thaliana Running head: GNAT family acetyltransferases in UV-B responses Julieta P. Fina and Paula Casati Centro de Estudios Fotosintéticos y Bioquímicos (CEFOBI), Universidad Nacional de Rosario, Suipacha 531, 2000 Rosario, Argentin Downloaded from Abbreviations: CBP, CREB-binding protein; ELP, ELongator complex Protein; GNAT, GCN5-related http://pcp.oxfordjournals.org/ N-acetyltransferase; GCN, General Control Nonderepressible protein; HAG, Histone acetyltransferase in the GNAT family; HAT, histone acetyltransferase; MS, Murashige and Skoog; RT–PCR; MYST, MOZ, Ybt2, Sas2, Tip60-like family proteins; reverse transcription–PCR; UTR, untranslated region; TAF, TATA binding proteing associated factor; UV, ultraviolet. by guest on October 27, 2016 2 ABSTRACT Histone acetylation is regulated by histone acetyltransferases and deacetylases. -
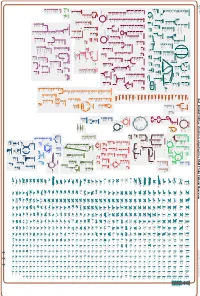
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000263195Cyc: Emticicia oligotrophica DSM 17448 Cellular Overview Connections between pathways are omitted for legibility. -

Biosynthesis of Fungal Alkyl Citrates and Polyketides
Biosynthesis of Fungal Alkyl Citrates and Polyketides Von der Naturwissenschaftlichen Fakultät Gottfried Wilhelm Leibniz Universität Hannover zur Erlangung des Grades Doktor der Naturwissenschaften (Dr. rer. nat.) genehmigte Dissertation von Sen Yin, Master (China) 2020 Referent: Prof. Dr. Russell Cox Korreferent: Prof. Dr. Andreas Kirschning Tag der Promotion: 02.10.2020 Abstract Abstract The main focus of the presented work concentrated on understanding the biosynthetic pathway of viridofungin, exploring the enzymatic properties of the key enzymes from the byssochlamic acid pathway, and engineering the PKS of tenellin. In a combined genetic and chemical approach, the biosynthetic pathway of viridiofungin and functions of enzymes from of byssochlamic acid pathway and programming of the tenellin PKS (TENS) system were elucidated. Trichoderma viride MF5628 was firstly genome sequenced by Illumina and Oxford nanopore sequencing. Then, using a combination of targeted gene knockout and RNA interference-based silencing in the native organism, a novel unreported tRNA ligase like enzyme involved in the biosynthetic pathway was identified. A citrate synthase like enzyme and a tRNA ligase like enzyme were found to be essential to the biosynthesis of viridiofungin. The biosynthesis of byssochlamic acid was investigated by protein expression and in vitro study. A hydrolase, citrate synthase, two 2-methyl citrate dehydratases, two KSI and two PEBP proteins were expressed in E. coli or yeast, in addition, in vtiro assay was carried out by using these proteins. During maleic anhydride monomer biosynthesis, a hydrolase can hydrolyze the hexaketide from ACP. Citrate synthase can use hexaketide CoA and oxaloacetate as substrates to form (2S, 3R) citrate. 2-Methyl citrate dehydratase takes (2S, 3R) citrate diasteroisomer as substrate to form a 2, 3-alkene product, and the product can spontaneously form a cyclised maleic anhydride monomer. -

A Genetic Selection System for Drug Discovery, Protein Engineering and Deciphering Biosynthetic Pathways
CHEMICAL COMPLEMENTATION: A GENETIC SELECTION SYSTEM FOR DRUG DISCOVERY, PROTEIN ENGINEERING AND DECIPHERING BIOSYNTHETIC PATHWAYS A Thesis Presented to The Academic Faculty By Bahareh Azizi In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy in the School of Chemistry and Biochemistry Georgia Institute of Technology August 2005 CHEMICAL COMPLEMENTATION: A GENETIC SELECTION SYSTEM FOR DRUG DISCOVERY, PROTEIN ENGINEERING AND DECIPHERING BIOSYNTHETIC PATHWAYS Approved By: Dr. Donald F. Doyle, Advisor Dr. Mostafa El-Sayed School of Chemistry and Biochemistry School of Chemistry and Biochemistry Georgia Institute of Technology Georgia Institute of Technology Dr. Sheldon May Dr. Jung Choi School of Chemistry and Biochemistry School of Biology Georgia Institute of Technology Georgia Institute of Technology Dr. Allen. Orville School of Chemistry and Biochemistry Date Approved: June 27, 2005 Georgia Institute of Technology In the land of repute, our passage they will dispute If this will not suit, don’t stay mute, and transform dictates of fate. When destitute and in need, let your love and passion breed Life’s alchemy, essence and seed, unimagined wealth shall create. - Persian Poet, Hafez, Ghazal 5 I dedicate this work to my parents, Mohammed Reza Azizi and Zahra Barez, for their hard work and sacrifices for my education. Thank you for your endless love and support, and all that you have taught me throughout my life. I could not have completed this work if it were not for your hard work and all of your love. Thank you for teaching me the beauty and value of an education, and for believing in me always.