Modular Program Report Disclaimer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ICD10 Diagnoses FY2018 AHD.Com
ICD10 Diagnoses FY2018 AHD.com A020 Salmonella enteritis A5217 General paresis B372 Candidiasis of skin and nail A040 Enteropathogenic Escherichia coli A523 Neurosyphilis, unspecified B373 Candidiasis of vulva and vagina infection A528 Late syphilis, latent B3741 Candidal cystitis and urethritis A044 Other intestinal Escherichia coli A530 Latent syphilis, unspecified as early or B3749 Other urogenital candidiasis infections late B376 Candidal endocarditis A045 Campylobacter enteritis A539 Syphilis, unspecified B377 Candidal sepsis A046 Enteritis due to Yersinia enterocolitica A599 Trichomoniasis, unspecified B3781 Candidal esophagitis A047 Enterocolitis due to Clostridium difficile A6000 Herpesviral infection of urogenital B3789 Other sites of candidiasis A048 Other specified bacterial intestinal system, unspecified B379 Candidiasis, unspecified infections A6002 Herpesviral infection of other male B380 Acute pulmonary coccidioidomycosis A049 Bacterial intestinal infection, genital organs B381 Chronic pulmonary coccidioidomycosis unspecified A630 Anogenital (venereal) warts B382 Pulmonary coccidioidomycosis, A059 Bacterial foodborne intoxication, A6920 Lyme disease, unspecified unspecified unspecified A7740 Ehrlichiosis, unspecified B387 Disseminated coccidioidomycosis A080 Rotaviral enteritis A7749 Other ehrlichiosis B389 Coccidioidomycosis, unspecified A0811 Acute gastroenteropathy due to A879 Viral meningitis, unspecified B399 Histoplasmosis, unspecified Norwalk agent A938 Other specified arthropod-borne viral B440 Invasive pulmonary -

Evaluation of the Uterine Causes of Female Infertility by Ultrasound: A
Evaluation of the Uterine Causes of Female Infertility by Ultrasound: A Literature Review Shohreh Irani (PhD)1, 2, Firoozeh Ahmadi (MD)3, Maryam Javam (BSc)1* 1 BSc of Midwifery, Department of Reproductive Imaging, Reproductive Biomedicine Research Center, Royan Institute for Reproductive Biomedicine, Iranian Academic Center for Education, Culture, and Research, Tehran, Iran 2 Assistant Professor, Department of Epidemiology and Reproductive Health, Reproductive Epidemiology Research Center, Royan Institute for Reproductive Biomedicine, Iranian Academic Center for Education, Culture, and Research, Tehran, Iran 3 Graduated, Department of Reproductive Imaging, Reproductive Biomedicine Research Center, Royan Institute for Reproductive Biomedicine, Iranian Academic Center for Education, Culture, and Research, Tehran, Iran A R T I C L E I N F O A B S T R A C T Article type: Background & aim: Various uterine disorders lead to infertility in women of Review article reproductive ages. This study was performed to describe the common uterine causes of infertility and sonographic evaluation of these causes for midwives. Article History: Methods: This literature review was conducted on the manuscripts published at such Received: 07-Nov-2015 databases as Elsevier, PubMed, Google Scholar, and SID as well as the original text books Accepted: 31-Jan-2017 between 1985 and 2015. The search was performed using the following keywords: infertility, uterus, ultrasound scan, transvaginal sonography, endometrial polyp, fibroma, Key words: leiomyoma, endometrial hyperplasia, intrauterine adhesion, Asherman’s syndrome, uterine Female infertility synechiae, adenomyosis, congenital uterine anomalies, and congenital uterine Menstrual cycle malformations. Ultrasound Results: A total of approximately 180 publications were retrieved from the Uterus respective databases out of which 44 articles were more related to our topic and studied as suitable references. -

Womens Health Requisition Forms
PCR - WHI Implementation PLACE ON SWAB 10854 Midwest Industrial Blvd. St. Louis, MO 63132 MM DD YY Phone: (314) 200-3040 | Fax (314) 200-3042 (1) CLIA ID #26D0953866 JANE DOE v3 PCR MOLECULAR REQUISITION - WOMEN'S HEALTH INFECTION PRACTICE INFORMATION PATIENT INFORMATION *SPECIMEN INFORMATION (2) DOE JANE MM/DD/YY LAST NAME FIRST NAME DATE COLLECTED W O M E ' N S H E A L T H I F N E C T I O N SSN MM/DD/YY HH:MM AM SSN DATE OF BIRTH TIME COLLECTED REQUESTING PHYSICIAN: DR. SWAB TESTER (3) Sex: F X M (4) Diagnosis Codes X N76.0 Acute vaginitis B37.49 Other urogenital candidiasis A54.9 Gonococcal infection, unspecified N76.1 Subacute and chronic vaginitis N89.8 Other specified noninflammatory disorders of vagina A59.00 Urogenital trichomoniasis, unspecified N76.2 Acute vulvitis O99.820 Streptococcus B carrier state complicating pregnancy A64 Unspecified sexually transmitted disease N76.3 Subacute and chronic vulvitis O99.824 Streptococcus B carrier state complicating childbirth A74.9 Chlamydial infection, unspecified N76.4 Abscess of vulva B95.1 Streptococcus, group B, as the cause Z11.3 Screening for infections with a predmoninantly N76.5 Ulceration of vagina of diseases classified elsewhere sexual mode of trasmission N76.6 Ulceration of vulva Z22.330 Carrier of group B streptococcus Other: N76.81 Mucositis(ulcerative) of vagina and vulva N70.91 Salpingitis, unspecified N76.89 Other specified inflammation of vagina and vulva N70.92 Oophoritis, unspecified N95.2 Post menopausal atrophic vaginitis N71.9 Inflammatory disease of uterus, unspecified -

(IJCRI) Abdominal Menstruation
www.edoriumjournals.com CASE SERIES PEER REVIEWED | OPEN ACCESS Abdominal menstruation: A dilemma for the gynecologist Seema Singhal, Sunesh Kumar, Yamini Kansal, Deepika Gupta, Mohit Joshi ABSTRACT Introduction: Menstrual fistulae are rare. They have been reported after pelvic inflammatory disease, pelvic radiation therapy, trauma, pelvic surgery, endometriosis, tuberculosis, gossypiboma, Crohn’s disease, sepsis, migration of intrauterine contraceptive device and other pelvic pathologies. We report two rare cases of menstrual fistula. Case Series: Case 1: A 27- year-old nulliparous female presented with complaint of cyclical bleeding from the abdomen since three years. There was previous history of hypomenorrhea and cyclical abdominal pain since menarche. There is history of laparotomy five years back and laparoscopy four years back in view of pelvic mass. Soon after she began to have blood mixed discharge from scar site which coincided with her menstruation. She was diagnosed to have a vertical fusion defect with communicating left hypoplastic horn and non-communicating right horn on imaging. Laparotomy with excision of fistula and removal of right hematosalpinx was done. Case 2: 25-year-old female presented with history of lower segment caesarean section (LSCS) and burst abdomen, underwent laparotomy and loop ileostomy. Thereafter patient developed cyclical bleeding from scar site. Laparotomy with excision of fistulous tract and closure of uterine rent was done. Conclusion: Clinical suspicion and imaging help to clinch the diagnosis. There is no recommended treatment modality. Surgery is the mainstay of management. Complete excision of fistulous tract is mandatory for good long-term outcomes. International Journal of Case Reports and Images (IJCRI) International Journal of Case Reports and Images (IJCRI) is an international, peer reviewed, monthly, open access, online journal, publishing high-quality, articles in all areas of basic medical sciences and clinical specialties. -

N35.12 Postinfective Urethral Stricture, NEC, Female N35.811 Other
N35.12 Postinfective urethral stricture, NEC, female N35.811 Other urethral stricture, male, meatal N35.812 Other urethral bulbous stricture, male N35.813 Other membranous urethral stricture, male N35.814 Other anterior urethral stricture, male, anterior N35.816 Other urethral stricture, male, overlapping sites N35.819 Other urethral stricture, male, unspecified site N35.82 Other urethral stricture, female N35.911 Unspecified urethral stricture, male, meatal N35.912 Unspecified bulbous urethral stricture, male N35.913 Unspecified membranous urethral stricture, male N35.914 Unspecified anterior urethral stricture, male N35.916 Unspecified urethral stricture, male, overlapping sites N35.919 Unspecified urethral stricture, male, unspecified site N35.92 Unspecified urethral stricture, female N36.0 Urethral fistula N36.1 Urethral diverticulum N36.2 Urethral caruncle N36.41 Hypermobility of urethra N36.42 Intrinsic sphincter deficiency (ISD) N36.43 Combined hypermobility of urethra and intrns sphincter defic N36.44 Muscular disorders of urethra N36.5 Urethral false passage N36.8 Other specified disorders of urethra N36.9 Urethral disorder, unspecified N37 Urethral disorders in diseases classified elsewhere N39.0 Urinary tract infection, site not specified N39.3 Stress incontinence (female) (male) N39.41 Urge incontinence N39.42 Incontinence without sensory awareness N39.43 Post-void dribbling N39.44 Nocturnal enuresis N39.45 Continuous leakage N39.46 Mixed incontinence N39.490 Overflow incontinence N39.491 Coital incontinence N39.492 Postural -

Recurrent Hematometra with Endometriosis in an Adolescent Girl: a Case Report
International Journal of Reproduction, Contraception, Obstetrics and Gynecology Garg R et al. Int J Reprod Contracept Obstet Gynecol. 2019 Nov;8(11):4567-4569 www.ijrcog.org pISSN 2320-1770 | eISSN 2320-1789 DOI: http://dx.doi.org/10.18203/2320-1770.ijrcog20194895 Case Report Recurrent hematometra with endometriosis in an adolescent girl: a case report Sarita Agrawal, Rajshree Sahu*, Pushpawati Thakur, Vinita Singh, Pawan B. Chandramohan Department of Obstetrics and Gynecology, All India Institute of Medical Sciences, Raipur, Chhattisgarh, India Received: 18 August 2019 Revised: 19 September 2019 Accepted: 09 October 2019 *Correspondence: Dr. Rajshree Sahu, E-mail: [email protected] Copyright: © the author(s), publisher and licensee Medip Academy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Hematometra is a collection or retention of blood in the uterine cavity. This condition is most commonly associated with congenital uterine anomalies that result from abnormal formation, fusion or resorption of Mullerian ducts during fetal life or may be due to prior surgical procedures, causing an obstruction of the genitourinary outflow tract. We report an unusual case of hematometra with endometriosis secondary to cervical stenosis. This is a rare and important case report due to the complexity of diagnosis as cervical stenosis was not presented as primary amenorrhoea as its usual presentation. This case was successfully managed by Hysteroscopic cervical dilatation under USG guidance followed by transcervical insertion of a catheter to prevent recurrent stenosis. -

Clinical Outcomes of Hysterectomy for Benign Diseases in the Female Genital Tract
Original article eISSN 2384-0293 Yeungnam Univ J Med 2020;37(4):308-313 https://doi.org/10.12701/yujm.2020.00185 Clinical outcomes of hysterectomy for benign diseases in the female genital tract: 6 years’ experience in a single institute Hyo-Shin Kim1, Yu-Jin Koo2, Dae-Hyung Lee2 1Department of Obstetrics and Gynecology, Yeungnam University Hospital, Daegu, Korea 2Department of Obstetrics and Gynecology, Yeungnam University College of Medicine, Daegu, Korea Received: March 17, 2020 Revised: April 7, 2020 Background: Hysterectomy is one of the major gynecologic surgeries. Historically, several surgical Accepted: April 14, 2020 procedures have been used for hysterectomy. The present study aims to evaluate the surgical trends and clinical outcomes of hysterectomy performed for benign diseases at the Yeungnam Corresponding author: University Hospital. Yu-Jin Koo Methods: We retrospectively reviewed patients who underwent a hysterectomy for benign dis- Department of Obstetrics and eases from 2013 to 2018. Data included the patients’ demographic characteristics, surgical indi- Gynecology, Yeungnam University cations, hysterectomy procedures, postoperative pathologies, and perioperative outcomes. College of Medicine, 170 Hyeonchung-ro, Nam-gu, Daegu Results: A total of 809 patients were included. The three major indications for hysterectomy were 42415, Korea uterine leiomyoma, pelvic organ prolapse, and adenomyosis. The most common procedure was Tel: +82-53-620-3433 total laparoscopic hysterectomy (TLH, 45.2%), followed by open hysterectomy (32.6%). During Fax: +82-53-654-0676 the study period, the rate of open hysterectomy was nearly constant (29.4%–38.1%). The mean E-mail: [email protected] operative time was the shortest in the single-port laparoscopic assisted vaginal hysterectomy (LAVH, 89.5 minutes), followed by vaginal hysterectomy (VH, 96.8 minutes) and TLH (105 min- utes). -

Gender Confirmation Surgery Reference Number: PA.CP.MP.95 Effective Date: 01/18 Coding Implications Last Review Date: 09/17 Revision Log
Clinical Policy: Gender Confirmation Surgery Reference Number: PA.CP.MP.95 Effective Date: 01/18 Coding Implications Last Review Date: 09/17 Revision Log Description Services for gender confirmation most often include hormone treatment, counseling, psychotherapy, complete hysterectomy, bilateral mastectomy, chest reconstruction or augmentation as appropriate, genital reconstruction, facial hair removal, and certain facial plastic reconstruction. Not every individual will require each intervention so necessity needs to be considered on an individualized basis. This criteria outlines medical necessity criteria for gender confirmation surgery when such services are included under the members’ benefit plan contract provisions. Policy/Criteria It is the policy of Pennsylvania Health and Wellness® (PHW) that the gender confirmation surgeries listed in section III are considered medically necessary for members when diagnosed with gender dysphoria per criteria in section I and when meeting eligibility criteria in section II. I. Gender Dysphoria Criteria, meets A and B A. Marked incongruence between the member’s experienced/expressed gender and assigned gender, of at least 6 month’s duration, as indicated by two or more of the following: 1. Marked incongruence between the member’s experienced/expressed gender and primary and/or secondary sex characteristics; 2. A strong desire to be rid of one’s primary and/or secondary sex characteristics because of a marked incongruence with one’s experienced/expressed gender; 3. A strong desire for the primary and/or secondary sex characteristics of the other gender; 4. A strong desire to be of the other gender (or some alternative gender different from one’s assigned gender); 5. A strong desire to be treated as the other gender (or some alternative gender different from one’s assigned gender); 6. -

Diagnostic Tests for Vaginosis/Vaginitis
Alberta Heritage Foundation for Medical Research Diagnostic tests for vaginosis/vaginitis Christa Harstall and Paula Corabian October 1998 HTA12 Diagnostic tests for vaginosis/vaginitis Christa Harstall and Paula Corabian October 1998 © Copyight Alberta Heritage Foundation for Medical Research, 1998 This Health Technology Assessment Report has been prepared on the basis of available information of which the Foundation is aware from public literature and expert opinion, and attempts to be current to the date of publication. The report has been externally reviewed. Additional information and comments relative to the Report are welcome, and should be sent to: Director, Health Technology Assessment Alberta Heritage Foundation for Medical Research 3125 Manulife Place, 10180 - 101 Street Edmonton Alberta T5J 3S4 CANADA Tel: 403-423-5727, Fax: 403-429-3509 This study is based, in part, on data provided by Alberta Health. The interpretation of the data in the report is that of the authors and does not necessarily represent the views of the Government of Alberta. ISBN 1-896956-15-7 Alberta's health technology assessment program has been established under the Health Research Collaboration Agreement between the Alberta Heritage Foundation for Medical Research and the Alberta Health Ministry. Acknowledgements The Alberta Heritage Foundation for Medical Research is most grateful to the following persons for their comments on the draft report and for provision of information. The views expressed in the final report are those of the Foundation. Dr. Jane Ballantine, Section of General Practice, Calgary Dr. Deirdre L. Church, Microbiology, Calgary Laboratory Services, Calgary Dr. Nestor N. Demianczuk, Royal Alexandra Hospital, Edmonton Dr. -
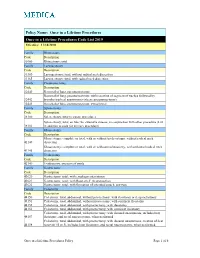
Once in a Lifetime Procedures Code List 2019 Effective: 11/14/2010
Policy Name: Once in a Lifetime Procedures Once in a Lifetime Procedures Code List 2019 Effective: 11/14/2010 Family Rhinectomy Code Description 30160 Rhinectomy; total Family Laryngectomy Code Description 31360 Laryngectomy; total, without radical neck dissection 31365 Laryngectomy; total, with radical neck dissection Family Pneumonectomy Code Description 32440 Removal of lung, pneumonectomy; Removal of lung, pneumonectomy; with resection of segment of trachea followed by 32442 broncho-tracheal anastomosis (sleeve pneumonectomy) 32445 Removal of lung, pneumonectomy; extrapleural Family Splenectomy Code Description 38100 Splenectomy; total (separate procedure) Splenectomy; total, en bloc for extensive disease, in conjunction with other procedure (List 38102 in addition to code for primary procedure) Family Glossectomy Code Description Glossectomy; complete or total, with or without tracheostomy, without radical neck 41140 dissection Glossectomy; complete or total, with or without tracheostomy, with unilateral radical neck 41145 dissection Family Uvulectomy Code Description 42140 Uvulectomy, excision of uvula Family Gastrectomy Code Description 43620 Gastrectomy, total; with esophagoenterostomy 43621 Gastrectomy, total; with Roux-en-Y reconstruction 43622 Gastrectomy, total; with formation of intestinal pouch, any type Family Colectomy Code Description 44150 Colectomy, total, abdominal, without proctectomy; with ileostomy or ileoproctostomy 44151 Colectomy, total, abdominal, without proctectomy; with continent ileostomy 44155 Colectomy, -

Page Mackup January-14.Qxd
Bangladesh Journal of Medical Science Vol. 13 No. 01 January’14 Case report: Unilateral Functional Uterine Horn with Non Functioning Rudimentary Horn and Cervico-Vaginal Agenesis: Case Report Hakim S1, Ahmad A2, Jain M3, Anees A4. ABSTRACT: Developmental anomalies involving Mullerian ducts are one of the most fascinating disorders in Gynaecology. The incidence rates vary widely and have been described between 0.1-3.5% in the general population. We report a case of a fifteen year old girl who presented with pri- mary amenorrhea and lower abdomen pain, with history of instrumentation about two months back. She was found to have abdominal lump of sixteen weeks size uterus. On examination vagina was found to be represented as a small blind pouch measuring 2-3cms in length. A rec- tovaginal fistula (2x2 cms) was also observed. Ultrasonography of abdomen revealed bulky uterus (size 11.2x6 cm) with 150 millilitre of collection. A diagnosis of hematometra with iatro- genic fistula was made. Vaginal drainage of hematometra was done which was followed by laparotomy. Peroperatively she was found to have a left side unicornuate uterus with right side small rudimentary horn. Left fallopian tube and ovary showed dense adhesions and multiple endometriotic implants. Both cervix and vagina were absent. Total abdominal hysterectomy was done and rectovaginal fistula repaired. The present case is reported due to its rarity as it involved both mullerian agenesis with cervical and vaginal agenesis along with disorder of lat- eral fusion. This is an asymmetric type of mullerian duct development in which arrest has occurred in different stages of development on two sides. -

Gynecological-DBQ
INTERNAL VETERANS AFFAIRS USE GYNECOLOGICAL CONDITIONS DISABILITY BENEFITS QUESTIONNAIRE IMPORTANT - THE DEPARTMENT OF VETERANS AFFAIRS (VA) WILL NOT PAY OR REIMBURSE ANY EXPENSES OR COST INCURRED IN THE PROCESS OF COMPLETING AND/OR SUBMITTING THIS FORM. PLEASE READ THE PRIVACY ACT AND RESPONDENT BURDEN INFORMATION ON REVERSE BEFORE COMPLETING FORM. NAME OF PATIENT/VETERAN PATIENT/VETERAN'S SOCIAL SECURITY NUMBER NOTE TO PHYSICIAN - Your patient is applying to the U.S. Department of Veterans Affairs (VA) for disability benefits. VA will consider the information you provide on this questionnaire as part of their evaluation in processing the claim. VA reserves the right to confirm the authenticity of ALL DBQs completed by private health care providers. IS THIS DBQ BEING COMPLETED IN CONJUNCTION WITH A VA21-2507, C&P EXAMINATION REQUEST? YES NO If no, how was the examination completed (check all that apply)? In-person examination Records reviewed Other, please specify: Comments: ACCEPTABLE CLINICAL EVIDENCE (ACE) INDICATE METHOD USED TO OBTAIN MEDICAL INFORMATION TO COMPLETE THIS DOCUMENT: Review of available records (without in-person or video telehealth examination) using the Acceptable Clinical Evidence (ACE) process because the existing medical evidence provided sufficient information on which to prepare the DBQ and such an examination will likely provide no additional relevant evidence. Review of available records in conjunction with a telephone interview with the Veteran (without in-person or telehealth examination) using the ACE process because the existing medical evidence supplemented with a telephone interview provided sufficient information on which to prepare the DBQ and such an examination would likely provide no additional relevant evidence.