Photochemistry of Polycyclic Aromatic Hydrocarbons in Cosmic Water Ice I
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And Abiogenesis
Historical Development of the Distinction between Bio- and Abiogenesis. Robert B. Sheldon NASA/MSFC/NSSTC, 320 Sparkman Dr, Huntsville, AL, USA ABSTRACT Early greek philosophers laid the philosophical foundations of the distinction between bio and abiogenesis, when they debated organic and non-organic explanations for natural phenomena. Plato and Aristotle gave organic, or purpose-driven explanations for physical phenomena, whereas the materialist school of Democritus and Epicurus gave non-organic, or materialist explanations. These competing schools have alternated in popularity through history, with the present era dominated by epicurean schools of thought. Present controversies concerning evidence for exobiology and biogenesis have many aspects which reflect this millennial debate. Therefore this paper traces a selected history of this debate with some modern, 20th century developments due to quantum mechanics. It ¯nishes with an application of quantum information theory to several exobiology debates. Keywords: Biogenesis, Abiogenesis, Aristotle, Epicurus, Materialism, Information Theory 1. INTRODUCTION & ANCIENT HISTORY 1.1. Plato and Aristotle Both Plato and Aristotle believed that purpose was an essential ingredient in any scienti¯c explanation, or teleology in philosophical nomenclature. Therefore all explanations, said Aristotle, answer four basic questions: what is it made of, what does it represent, who made it, and why was it made, which have the nomenclature material, formal, e±cient and ¯nal causes.1 This aristotelean framework shaped the terms of the scienti¯c enquiry, invisibly directing greek science for over 500 years. For example, \organic" or \¯nal" causes were often deemed su±cient to explain natural phenomena, so that a rock fell when released from rest because it \desired" its own kind, the earth, over unlike elements such as air, water or ¯re. -

Laboratory Studies for Planetary Sciences
Laboratory Studies for Planetary Sciences A Planetary Decadal Survey White Paper Prepared by the American Astronomical Society (AAS) Working Group on Laboratory Astrophysics (WGLA) http://www.aas.org/labastro Targeted Panels: Terrestrial Planets; Outer Solar System Satellites; Small Bodies Lead Author: Murthy S. Gudipati Ice Spectroscopy Lab, Science Division, Mail Stop 183-301, Jet Propulsion Laboratory, California Institute of Technology, 4800 Oak Grove Drive, Pasadena, CA 91109. [email protected], 818-354-2637 Co-Authors: Michael A'Hearn - University of Maryland [email protected], 301-405-6076 Nancy Brickhouse - Harvard-Smithsonian Center for Astrophysics [email protected], 617-495-7438 John Cowan - University of Oklahoma [email protected], 405-325-3961 Paul Drake - University of Michigan [email protected], 734-763-4072 Steven Federman - University of Toledo [email protected], 419-530-2652 Gary Ferland - University of Kentucky [email protected], 859-257-879 Adam Frank - University of Rochester [email protected], 585-275-1717 Wick Haxton - University of Washington [email protected], 206-685-2397 Eric Herbst - Ohio State University [email protected], 614-292-6951 Michael Mumma - NASA/GSFC [email protected], 301-286-6994 Farid Salama - NASA/Ames Research Center [email protected], 650-604-3384 Daniel Wolf Savin - Columbia University [email protected], 212-854-4124, Lucy Ziurys – University of Arizona [email protected], 520-621-6525 1 Brief Description: The WGLA of the AAS promotes collaboration and exchange of knowledge between astronomy and planetary sciences and the laboratory sciences (physics, chemistry, and biology). -

Viscosity of Gases References
VISCOSITY OF GASES Marcia L. Huber and Allan H. Harvey The following table gives the viscosity of some common gases generally less than 2% . Uncertainties for the viscosities of gases in as a function of temperature . Unless otherwise noted, the viscosity this table are generally less than 3%; uncertainty information on values refer to a pressure of 100 kPa (1 bar) . The notation P = 0 specific fluids can be found in the references . Viscosity is given in indicates that the low-pressure limiting value is given . The dif- units of μPa s; note that 1 μPa s = 10–5 poise . Substances are listed ference between the viscosity at 100 kPa and the limiting value is in the modified Hill order (see Introduction) . Viscosity in μPa s 100 K 200 K 300 K 400 K 500 K 600 K Ref. Air 7 .1 13 .3 18 .5 23 .1 27 .1 30 .8 1 Ar Argon (P = 0) 8 .1 15 .9 22 .7 28 .6 33 .9 38 .8 2, 3*, 4* BF3 Boron trifluoride 12 .3 17 .1 21 .7 26 .1 30 .2 5 ClH Hydrogen chloride 14 .6 19 .7 24 .3 5 F6S Sulfur hexafluoride (P = 0) 15 .3 19 .7 23 .8 27 .6 6 H2 Normal hydrogen (P = 0) 4 .1 6 .8 8 .9 10 .9 12 .8 14 .5 3*, 7 D2 Deuterium (P = 0) 5 .9 9 .6 12 .6 15 .4 17 .9 20 .3 8 H2O Water (P = 0) 9 .8 13 .4 17 .3 21 .4 9 D2O Deuterium oxide (P = 0) 10 .2 13 .7 17 .8 22 .0 10 H2S Hydrogen sulfide 12 .5 16 .9 21 .2 25 .4 11 H3N Ammonia 10 .2 14 .0 17 .9 21 .7 12 He Helium (P = 0) 9 .6 15 .1 19 .9 24 .3 28 .3 32 .2 13 Kr Krypton (P = 0) 17 .4 25 .5 32 .9 39 .6 45 .8 14 NO Nitric oxide 13 .8 19 .2 23 .8 28 .0 31 .9 5 N2 Nitrogen 7 .0 12 .9 17 .9 22 .2 26 .1 29 .6 1, 15* N2O Nitrous -

Infrared Spectroscopy of Matrix-Isolated Polycyclic Aromatic Hydrocarbon Ions
J. Phys. Chem. A 2000, 104, 3655-3669 3655 Infrared Spectroscopy of Matrix-Isolated Polycyclic Aromatic Hydrocarbon Ions. 5. PAHs Incorporating a Cyclopentadienyl Ring† D. M. Hudgins,* C. W. Bauschlicher, Jr., and L. J. Allamandola NASA Ames Research Center, MS 245-6, Moffett Field, California 94035 J. C. Fetzer CheVron Research Company, Richmond, California 94802 ReceiVed: NoVember 5, 1999; In Final Form: February 9, 2000 The matrix-isolation technique has been employed to measure the mid-infrared spectra of the ions of several polycyclic aromatic hydrocarbons whose structures incorporate a cyclopentadienyl ring. These include the cations of fluoranthene (C16H10), benzo[a]fluoranthene, benzo[b]fluoranthene, benzo[j]fluoranthene, and benzo- [k]fluoranthene (all C20H12 isomers), as well as the anions of benzo[a]fluoranthene and benzo[j]fluoranthene. With the exception of fluoranthene, which presented significant theoretical difficulties, the experimental data are compared to theoretically calculated values obtained using density functional theory (DFT) at the B3LYP/ 4-31G level. In general, there is good overall agreement between the two data sets, with the positional agreement between the experimentally measured and theoretically predicted bands somewhat better than that associated with their intensities. The results are also consistent with previous experimental studies of polycyclic aromatic hydrocarbon ions. Specifically, in both the cationic and anionic species the strongest ion bands typically cluster in the 1450 to 1300 cm-1 range, reflecting an order-of-magnitude enhancement in the CC stretching and CH in-plane bending modes between 1600 and 1100 cm-1 in these species. The aromatic CH out-of- plane bending modes, on the other hand, are usually modestly suppressed (e 2x - 5x) in the cations relative to those of the neutral species, with the nonadjacent CH modes most strongly affected. -

Measurement of Cp/Cv for Argon, Nitrogen, Carbon Dioxide and an Argon + Nitrogen Mixture
Measurement of Cp/Cv for Argon, Nitrogen, Carbon Dioxide and an Argon + Nitrogen Mixture Stephen Lucas 05/11/10 Measurement of Cp/Cv for Argon, Nitrogen, Carbon Dioxide and an Argon + Nitrogen Mixture Stephen Lucas With laboratory partner: Christopher Richards University College London 5th November 2010 Abstract: The ratio of specific heats, γ, at constant pressure, Cp and constant volume, Cv, have been determined by measuring the oscillation frequency when a ball bearing undergoes simple harmonic motion due to the gravitational and pressure forces acting upon it. The γ value is an important gas property as it relates the microscopic properties of the molecules on a macroscopic scale. In this experiment values of γ were determined for input gases: CO2, Ar, N2, and an Ar + N2 mixture in the ratio 0.51:0.49. These were found to be: 1.1652 ± 0.0003, 1.4353 ± 0.0003, 1.2377 ± 0.0001and 1.3587 ± 0.0002 respectively. The small uncertainties in γ suggest a precise procedure while the discrepancy between experimental and accepted values indicates inaccuracy. Systematic errors are suggested; however it was noted that an average discrepancy of 0.18 between accepted and experimental values occurred. If this difference is accounted for, it can be seen that we measure lower vibrational contributions to γ at room temperature than those predicted by the equipartition principle. It can be therefore deduced that the classical idea of all modes contributing to γ is incorrect and there is actually a „freezing out‟ of vibrational modes at lower temperatures. I. Introduction II. Method The primary objective of this experiment was to determine the ratio of specific heats, γ, for gaseous Ar, N2, CO2 and an Ar + N2 mixture. -

Liquid Argon
Safetygram 8 Liquid argon Liquid argon is tasteless, colorless, odorless, noncorrosive, nonflammable, and extremely cold. Belonging to the family of rare gases, argon is the most plentiful, making up approximately 1% of the earth’s atmosphere. It is monatomic and extremely inert, forming no known chemical compounds. Since argon is inert, special materials of construction are not required. However, materials of construction must be selected to withstand the low temperature of liquid argon. Vessels and piping should be designed to American Society of Mechanical Engineers (ASME) specifications or the Department of Transportation (DOT) codes for the pressures and temperatures involved. Although used more commonly in the gaseous state, argon is commonly stored and transported as a liquid, affording a more cost-effective way of providing product supply. Liquid argon is a cryogenic liquid. Cryogenic liquids are liquefied gases that have a normal boiling point below –130°F (–90°C). Liquid argon has a boiling point of –303°F (–186°C). The temperature difference between the product and the surrounding environment, even in winter, is substantial. Keeping this surrounding heat from the product requires special equipment to store and handle cryogenic liquids. A typical system consists of the following components: a cryogenic storage tank, one or more vaporizers, a pressure control system, and all of the piping required for fill, vaporization. The cryogenic tank is constructed, in principle, like a vacuum bottle. It is designed to keep heat away from the liquid that is contained in the inner vessel. Vaporizers convert the liquid argon to its gaseous state. A pressure control manifold controls the pressure at which the gas is fed to the process. -

Absorption and Laser Induced Fluorescence Spectroscopy Of
Astronomy in Focus, Volume 1, Focus Meeting 12, E17 XXIXth IAU General Assembly, August 2015 c International Astronomical Union 2017 Piero Benvenuti, ed. doi:10.1017/S1743921317005075 Absorption and Laser Induced Fluorescence Spectroscopy of Neutral Polycyclic Aromatic Hydrocarbons in Argon matrices Salma Bejaoui 1,2, Farid Salama 1 and Ella Sciamma-O’Brien 1,3 1 NASA Ames Research Center, Mail Stop 245-6, Moffett Field, California 94035-1000 2 NPP, Oak Ridge Associated Universities 3 BAER Institute, Petaluma, CA email: [email protected] Keywords. PAH, fluorescence, UV-VIS, ERE, comet. Polycyclic aromatic hydrocarbons (PAHs) are considered as plausible carriers for the extended red emission (ERE), a photoluminescent process associated with a wide variety of interstellar environments, as well as for broad emission band features seen in cometary spectra. We report the absorption spectra of phenanthrene, anthracene, fluoranthene, pentacene, pyrene, chrysene and triphenylene isolated at 10 K in solid argon matrices together with laser induced fluores- cence (LIF) spectra at 355 nm of matrix-isolated anthracene and fluoranthene. LIF spectra are compared with the UV/blue fluorescence spectra of the Red Rectangle Nebula (RR). The LIF spectra measured in solid Ar matrices have been shifted to the predicted position of the PAH band emission in the gas phase for comparison with the astronomical observations (Fig. 1). These preliminary results indicate that small neutral PAHs can well account for the blue fluorescence observed in the RR as it has been previously proposed (Vijh, et al. (2004)). LIF spectra of anthracene measured in Ar matrices are also compared to the emission spectra of 1P/Halley’s inner coma(Fig. -

Computational Surface Modelling of Ices and Minerals of Interstellar Interest—Insights and Perspectives
minerals Review Computational Surface Modelling of Ices and Minerals of Interstellar Interest—Insights and Perspectives Albert Rimola 1,* , Stefano Ferrero 1, Aurèle Germain 2 , Marta Corno 2 and Piero Ugliengo 2,3 1 Departament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Catalonia, Spain; [email protected] 2 Dipartimento di Chimica, Università degli Studi di Torino, 10125 Torino, Italy; [email protected] (A.G.); [email protected] (M.C.); [email protected] (P.U.) 3 Nanostructured Interfaces and Surfaces (NIS) Centre, Università degli Studi di Torino, 10125 Torino, Italy * Correspondence: [email protected]; Tel.: +34-93-581-3723 Abstract: The universe is molecularly rich, comprising from the simplest molecule (H2) to complex organic molecules (e.g., CH3CHO and NH2CHO), some of which of biological relevance (e.g., amino acids). This chemical richness is intimately linked to the different physical phases forming Solar-like planetary systems, in which at each phase, molecules of increasing complexity form. Interestingly, synthesis of some of these compounds only takes place in the presence of interstellar (IS) grains, i.e., solid-state sub-micron sized particles consisting of naked dust of silicates or carbonaceous materials that can be covered by water-dominated ice mantles. Surfaces of IS grains exhibit particular characteristics that allow the occurrence of pivotal chemical reactions, such as the presence of binding/catalytic sites and the capability to dissipate energy excesses through the grain phonons. The present know-how on the physicochemical features of IS grains has been obtained by the fruitful synergy of astronomical observational with astrochemical modelling and laboratory experiments. -

Interstellar Dust Within the Life Cycle of the Interstellar Medium K
EPJ Web of Conferences 18, 03001 (2011) DOI: 10.1051/epjconf/20111803001 C Owned by the authors, published by EDP Sciences, 2011 Interstellar dust within the life cycle of the interstellar medium K. Demyk1,2,a 1Université de Toulouse, UPS-OMP, IRAP, Toulouse, France 2CNRS, IRAP, 9 Av. colonel Roche, BP. 44346, 31028 Toulouse Cedex 4, France Abstract. Cosmic dust is omnipresent in the Universe. Its presence influences the evolution of the astronomical objects which in turn modify its physical and chemical properties. The nature of cosmic dust, its intimate coupling with its environment, constitute a rich field of research based on observations, modelling and experimental work. This review presents the observations of the different components of interstellar dust and discusses their evolution during the life cycle of the interstellar medium. 1. INTRODUCTION Interstellar dust grains are found everywhere in the Universe: in the Solar System, around stars at all evolutionary stages, in interstellar clouds of all kind, in galaxies and in the intergalactic medium. Cosmic dust is intimately mixed with the gas-phase and represents about 1% of the gas (in mass) in our Galaxy. The interstellar extinction and the emission of diffuse interstellar clouds is reproduced by three dust components: a population of large grains, the BGs (Big Grains, ∼10–500 nm) made of silicate and a refractory mantle, a population of carbonaceous nanograins, the VSGs (Very Small Grains, 1–10 nm) and a population of macro-molecules the PAHs (Polycyclic Aromatic Hydrocarbons) [1]. These three components are more or less abundant in the diverse astrophysical environments reflecting the coupling of dust with the environment and its evolution according to the physical and dynamical conditions. -

Hydrogen, Nitrogen and Argon Chemistry
Hydrogen, Nitrogen and Argon Chemistry Author: Brittland K. DeKorver Institute for Chemical Education and Nanoscale Science and Engineering Center University of Wisconsin-Madison Purpose: To learn about 3 of the gases that are in Earth’s atmosphere. Learning Objectives: 1. Understand that the atmosphere is made up of many gases. 2. Learn about the differences in flammability of nitrogen and hydrogen. 3. Learn what is produced during electrolysis. Next Generation Science Standards (est. 2013): PS1.A: Structure and Properties of Matter PS1.B: Chemical Reactions PS3.D: Energy in Chemical Processes and Everyday Life (partial) ETS1.A: Defining Engineering Problems ETS1.B: Designing Solutions to Engineering Problems ETS1.C: Optimizing the Design Solution National Science Education Standards (valid 1996-2013): Physical Science Standards: Properties and changes of properties in matter Earth and Space Science Standards: Structure of the earth system Suggested Previous Activities: Oxygen Investigation and Carbon Dioxide Chemistry Grade Level: 2-8 Time: 1 hour Materials: Safety glasses Manganese dioxide Work gloves (for handling Tea-light candles steel wool) Matches Test tubes Spatulas Beakers Wooden splints 100mL graduated cylinders Waste bucket for burnt 15mL graduated cylinders materials 3% hydrogen peroxide Vinegar Baking soda Safety: 1. Students should wear safety glasses during all activities. 2. Students should not be allowed to use matches or candle. 3. Students should be cautioned to only mix the chemicals as directed. Introduction: Air refers to the mixture of gases that make up the Earth’s atmosphere. In this lesson, students will learn about hydrogen (present in very, very small amounts in our atmosphere), nitrogen (the most prevalent gas), and argon (third behind nitrogen and oxygen.) Procedures: 1. -

Interstellar Ice Photochemistry and the Deuterium Enrichment and Chemical Properties of Meteoritic Polycyclic Aromatic Hydrocarbons S
63rd Annual Meteoritical Society Meeting 5003.pdf INTERSTELLAR ICE PHOTOCHEMISTRY AND THE DEUTERIUM ENRICHMENT AND CHEMICAL PROPERTIES OF METEORITIC POLYCYCLIC AROMATIC HYDROCARBONS S. A. Sandford1, M. P. Bernstein1,2, L. J. Allamandola1, J. S. Gillette3, and R. N. Zare3, 1NASA-Ames Research Center, MS 245-6, Moffett Field CA 94035 ([email protected]), 2SETI Institute, 2035 Landings Drive, Mountain View CA 94043, USA, 3Department of Chemistry, Stanford University, Stanford CA 94305, USA. Introduction: Polycyclic aromatic hydrocarbons surroundings over time. However, exchanged D atoms (PAHs) are known to be abundant and ubiquitous in at the other sites are labile only under UV irradiation; the interstellar medium (ISM) [1]. At the temperatures once irradiation ceases these atoms will not exchange typical of dense interstellar molecular clouds (T = 10–- with the environment. 50 K), the birth sites of stars and planetary systems, Because the aromatic fractions of meteoritic PAHs will be frozen out of the gas phase into icy grain organics are known to be enriched in deuterium [6], mantles. These mantles contain a number of molecular this raises the possibility that interstellar species, the most abundant being H2O, CH3OH, CO, photoprocessing of PAHs in D-enriched interstellar CO2, NH3, CH 4, and possibly N2 and O2 [2]. Many of ices has occurred [7]. The measured D/H ratios of these molecules are expected to be enriched in interstellar ices [8] and the observed exchange rates for deuterium via a number of interstellar chemical coronene (C 24H12)-D2O and d12-coronene (C24D12)-H2O processes [2]. While frozen in these D-enriched ices, isotopic substitution experiments show that PAHs in the PAHs will be exposed to ultraviolet photons from interstellar ices could easily attain the D/H levels nearby stars, the attenuated diffuse ISM radiation field, observed in meteorites [7]. -
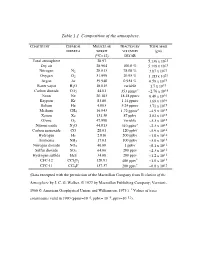
Table 1.1 Composition of the Atmosphere
Table 1.1 Composition of the atmosphere. CONSTITUENT CHEMICAL MOLECULAR FRACTION BY TOTAL MASS FORMULA WEIGHT VOLUME IN (gm) (12C=12) DRY AIR Total atmosphere 28.97 5.136 x 1021 Dry air 28.964 100.0 % 5.119 x 1021 Nitrogen N2 28.013 78.08 % 3.87 x 1021 Oxygen O2 31.999 20.95 % 1.185 x 1021 Argon Ar 39.948 0.934 % 6.59 x 1019 Water vapor H2O 18.015 variable 1.7 x 1019 Carbon dioxide CO2 44.01 353 ppmv* ~2.76 x 1018 Neon Ne 20.183 18.18 ppmv 6.48 x 1016 Krypton Kr 83.80 1.14 ppmv 1.69 x 1016 Helium He 4.003 5.24 ppmv 3.71 x 1015 Methane CH4 16.043 1.72 ppmv* ~4.9 x 1015 Xenon Xe 131.30 87 ppbv 2.02 x 1015 Ozone O3 47.998 variable ~3.3 x 1015 Nitrous oxide N2O 44.013 310 ppbv* ~2.3 x 1015 Carbon monoxide CO 28.01 120 ppbv ~5.9 x 1014 Hydrogen H2 2.016 500 ppbv ~1.8 x 1014 Ammonia NH3 17.03 100 ppbv ~3.0 x 1013 Nitrogen dioxide NO2 46.00 1 ppbv ~8.1 x 1012 Sulfur dioxide SO2 64.06 200 pptv ~2.3 x 1012 Hydrogen sulfide H2S 34.08 200 pptv ~1.2 x 1012 * 13 CFC-12 CCl2F2 120.91 480 pptv ~1.0 x 10 * 12 CFC-11 CCl3F 137.37 280 pptv ~6.8 x 10 (Data excerpted with the permission of the Macmillan Company from Evolution of the Atmosphere by J.