Differential Expression of APE1 and APE2 in Germinal Centers Promotes Error-Prone Repair and A:T Mutations During Somatic Hypermutation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Immunodeficiency Patients Differentiation of Common Variable
Defined Blocks in Terminal Plasma Cell Differentiation of Common Variable Immunodeficiency Patients This information is current as Nadine Taubenheim, Marcus von Hornung, Anne Durandy, of September 24, 2021. Klaus Warnatz, Lynn Corcoran, Hans-Hartmut Peter and Hermann Eibel J Immunol 2005; 175:5498-5503; ; doi: 10.4049/jimmunol.175.8.5498 http://www.jimmunol.org/content/175/8/5498 Downloaded from References This article cites 35 articles, 13 of which you can access for free at: http://www.jimmunol.org/content/175/8/5498.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 24, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Defined Blocks in Terminal Plasma Cell Differentiation of Common Variable Immunodeficiency Patients1 Nadine Taubenheim,* Marcus von Hornung,* Anne Durandy,† Klaus Warnatz,‡ Lynn Corcoran,§ Hans-Hartmut Peter,‡ and Hermann Eibel2* Common variable immunodeficiency (CVID) is a heterogeneous disorder characterized by defective Ab production and recurrent bacterial infections. -

Evaluation of the Effects of Activation Dependent Regulation of Cd40l on Germinal Center Response Using Cd40lδ5 Mouse Model
EVALUATION OF THE EFFECTS OF ACTIVATION DEPENDENT REGULATION OF CD40L ON GERMINAL CENTER RESPONSE USING CD40LΔ5 MOUSE MODEL By BITHA NARAYANAN A dissertation submitted to the School of Graduate Studies Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Microbiology and Molecular Genetics written under the direction of Lori R. Covey and approved by _____________________________________ _____________________________________ _____________________________________ _____________________________________ New Brunswick, New Jersey January 2020 ©[2020] Bitha Narayanan ALL RIGHTS RESERVED ABSTRACT OF THE DISSERTATION ACTIVATION-DEPENDENT POSTTRANSCRIPTIONAL REGULATION OF CD40L IS REQUIRED FOR AN OPTIMAL GERMINAL CENTER (GC) RESPONSE. by BITHA NARAYANAN Dissertation Director Dr. Lori R. Covey The interaction between cognate T and B cells decides the progression of an immune response to a pathogen or self-antigen. Of the multiple signals that synchronize to fine-tune this union, the binding of CD40 on the surface of B cells to CD40L expressed on CD4 T cells is of paramount importance. Ligation of CD40 on antigen- experienced B cells is associated with the initiation and development of germinal centers (GCs) resulting in the subsequent generation of high affinity antibodies and B cell memory. Post-transcriptional regulation of CD40L has been implicated in regulating the activation dependent expression of this protein. Our lab has previously shown a polypyrimidine tract binding protein complex binds to the 3’ UTR of the CD40L mRNA ii and that the deletion of a PTBP1 binding stability element in the same results in a significant decrease in the half-life of the CD40L transcript and subsequently the surface expression at later stages of activation in vitro. -

Does Recycling in Germinal Centers Exist?
Does Recycling in Germinal Centers Exist? Michael Meyer-Hermann Institut f¨ur Theoretische Physik, TU Dresden, D-01062 Dresden, Germany E-Mail: [email protected] Abstract: A general criterion is formulated in order to decide if recycling of B-cells exists in GC reactions. The criterion is independent of the selection and affinity maturation process and based solely on total centroblast population arguments. An experimental test is proposed to verify whether the criterion is fulfilled. arXiv:physics/0102062v2 [physics.bio-ph] 6 Mar 2002 1 1 Introduction The affinity maturation process in germinal center (GC) reactions has been well charac- terized in the last decade. Despite a lot of progress concerning the morphology of GCs and the stages of the selection process, a fundamental question remains unsolved: Does recycling exist? Recycling means a back differentiation of antibody presenting centrocytes (which undergo the selection process and interact with antigen fragments or T-cells) to centroblasts (which do not present antibodies and therefore do not interact with antigen fragments but proliferate and mutate). The existence of recycling in the GC has important consequences for the structure of the affinity maturation process in GC reactions. The centroblasts proliferate and mutate with high rates in the environment of follicular dendritic cells. During the GC reaction they differentiate to antibody presenting centrocytes which may then be selected by inter- action with antigen fragments and T-cells. If the positively selected centrocytes recycle to (proliferating and mutating) centroblasts the antibody-optimization process in GCs may be compatible with random mutations of the centroblasts. -

In Situ Observation of Germinal Center Cell Apoptosis During a Secondary Immune Response
J Clin Exp Hematopathol Vol. 46, No. 2, Nov 2006 Original Article In Situ Observation of Germinal Center Cell Apoptosis During a Secondary Immune Response Hito-aki Saitoh, Kunihiko Maeda, and Mitsunori Yamakawa Germinal centers are highly organized anatomic structures essential for the clonal expansion of germinal center (GC) B- cells and associated somatic hypermutation, isotype switching, selection of the high-affinity B-cells (affinity maturation), and elimination of irrelevant or autoreactive clones. The identification of cellular interactions and regulatory mechanisms controlling apoptosis within GCs is essential for a complete understanding of the cellular and molecular dynamics of the GC reaction. We performed a kinetic analysis of the apoptotic activity occurring within GCs of draining lymph nodes of mice immunized with sheep red blood cells (SRBC) after secondary stimulation. The apoptotic activity of GC cells can be divided into three distinct phases : 1) initial phase (within the first days after immunization), 2) reactive phase (from the 5th day to 15th day after secondary immunization), and 3) late phase (after the 15th day). Apoptosis decreased shortly after secondary immunization followed by an increase to peak after an additional 10 days. Finally, apoptosis of GC cells decreased to basal levels. Administration of apoptosis inhibitors decreased the amount of apoptosis during the reactive phase. These results suggest that the reactive phase may be the critical period in which clonal selection and cellular differentiation -
Lymph Node Cytology
Lymph node cytopathology : A practical approach to lymphoproliferative disorders • Koray Ceyhan, M.D • Department of Pathology • Faculty of Medicine • Ankara University • Ankara, Turkey Diagnostic use of FNA in lymph node pathologies • Well-established : • - metastatic malignancy, • - lymphoma recurrences • -some reactive or inflammatory disorders: Tuberculosis,sarcoidosis • Diagnostic sensitivity/accuracy : usually above 95% • Controversial: • primary lymphoma diagnosis • Diagnostic sensitivity varies from 12% to 96% Academic institutions: high level diagnostic accuracy Community practise the accuracy rate significantly low Multiparameter approach is critical for definitive lymphoma diagnosis • Cytomorphologic features alone are not sufficient for the diagnosis of primary lymphoma • Immunophenotyping with flow cytometry and/or immunocytochemistry is mandatory • In selected cases molecular/cytogenetic analyses are required for definitive lymphoma classification Lymph node pathologies • 1-Reactive lymphoid hyperplasia/inflammatory disorders • 2-Lymphoid malignancies • 3-Metastatic tumors 1 3 2 Common problems in lymph node cytology • Reactive lymphoid hyperplasia vs lymphoma • Primary lymphoma diagnosis(lymphoma subtyping) • Predicting primary site of metastatic tumor • Nonlymphoid tumors mimicking lymphoid malignancies • Correct diagnosis of specific benign lymphoid lesions Problem 1: Reactive vs lymphoma Case 19- years-old boy Multiple bilateral cervical LAPs for 4 weeks FNA from the largest cervical lymph node measuring 15X13 mm No -

MUM1: a Step Ahead Toward the Understanding of Lymphoma Histogenesis G Gaidano1 and a Carbone2
Leukemia (2000) 14, 563–566 2000 Macmillan Publishers Ltd All rights reserved 0887-6924/00 $15.00 www.nature.com/leu EDITORIAL MUM1: a step ahead toward the understanding of lymphoma histogenesis G Gaidano1 and A Carbone2 1Division of Internal Medicine, Department of Medical Sciences, Amedeo Avogadro University of Eastern Piedmont, Novara, and 2Division of Pathology, Centro di Riferimento Oncologico, IRCCS, Istituto Nazionale Tumori, Aviano, Italy In recent times, the field of B cell lymphoma histogenesis has togenesis has progressed rapidly due to the increasing avail- progressed rapidly due to the increasing availability of histo- ability of histogenetic markers of lymphoid cells.2 In normal genetic markers. Genotypic markers of B cell histogenesis are represented by mutations of IgV and BCL-6 genes, which are B cell physiology, the presence, or absence, of these histogen- somatically acquired at the time of B cell transit through the etic markers denotes different stages of lymphoid differen- germinal center (GC). Phenotypic markers are represented by tiation, allowing the distinction of mature B cells into virgin BCL-6 and CD138/syndecan-1 protein expression and allow the (ie pre-germinal center) B cells, germinal center B cells, and distinction between GC and post-GC B cells. On this basis, lym- post-germinal center (ie either memory B cells or plasmacells) phomas may be histogenetically distinguished into: (1) lym- B cells2 (Figure 2). Because these histogenetic markers are also phomas devoid of somatic IgV and BCL-6 hypermutation, which derive from pre-germinal center B cells; (2) lymphomas retained upon neoplastic transformation, the origin and differ- associated with somatic IgV and/or BCL-6 hypermutation and entiation stage of a given lymphoma may be temptatively BCL-6 expression, which closely reflect germinal center B assigned based on the combination of histogenetic markers cells; and (3) lymphomas associated with somatic IgV and/or associated with the tumor clone (Figure 2). -

The Broad Landscape of Follicular Lymphoma: Part I
PATHOLOGICA 2020;112:1-16; DOI: 10.32074/1591-951X-35-19 Review The broad landscape of follicular lymphoma: Part I Stefano Fratoni1, Magda Zanelli2, Maurizio Zizzo3,4, Francesca Sanguedolce5, Valentina Aimola6, Giulia Cerrone6, Linda Ricci7, Alessandra Filosa8, Giovanni Martino9, Stefano Ascani10 1 Department of Anatomic Pathology, St. Eugenio Hospital of Rome, Rome, Italy; 2 Pathology Unit, Azienda Unità Sanitaria Locale - IRCCS di Reggio Emilia, Italy; 3 Surgical Oncology Unit, Azienda Unità Sanitaria Locale - IRCCS di Reggio Emilia, Italy; 4 Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, Modena, Italy; 5 Pathology Unit, Azienda Ospedaliero-Universitaria - Ospedali Riuniti di Foggia, Italy; 6 Pathology Unit, University of Cagliari, Italy; 7 Pathology Unit, University of Siena, Italy; 8 Pathology Unit, Ospedale di Ascoli Piceno, Ascoli, Italy; 9 Hematology Unit, CREO, Azienda Ospedaliera di Perugia, University of Perugia, Italy; 10 Pathology Unit, Azienda Ospedaliera Santa Maria Terni, University of Perugia, Terni, Italy Summary Follicular lymphoma is a neoplasm derived from follicle center B cells, typically both cen- trocytes and centroblasts, in variable proportions according to the lymphoma grading. The pattern of growth may be entirely follicular, follicular and diffuse, and rarely completely dif- fuse. It represents the second most common non-Hodgkin lymphoma, after diffuse large B-cell lymphoma and is the most common low-grade mature B-cell lymphoma in western countries. In the majority of cases, follicular lymphoma is a nodal tumor, occurring in adults and frequently associated with the translocation t(14;18)(q32;q21)/IGH-BCL2. However, in recent years the spectrum of follicular lymphoma has expanded and small subsets of follicular lymphoma, which differ from common follicular lymphoma, have been identified Received and accepted: December 8, 2019 and included in the current 2017 WHO classification. -

B Cell–Intrinsic Mtorc1 Promotes Germinal Center–Defining
B Cell−Intrinsic mTORC1 Promotes Germinal Center−Defining Transcription Factor Gene Expression, Somatic Hypermutation, and Memory B Cell This information is current as Generation in Humoral Immunity of September 26, 2021. Ariel L. Raybuck, Sung Hoon Cho, Jingxin Li, Meredith C. Rogers, Keunwook Lee, Christopher L. Williams, Mark Shlomchik, James W. Thomas, Jin Chen, John V. Williams and Mark R. Boothby Downloaded from J Immunol published online 12 March 2018 http://www.jimmunol.org/content/early/2018/03/11/jimmun ol.1701321 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2018/03/11/jimmunol.170132 Material 1.DCSupplemental Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 26, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published March 12, 2018, doi:10.4049/jimmunol.1701321 The Journal of Immunology B Cell–Intrinsic mTORC1 Promotes Germinal Center–Defining Transcription Factor Gene Expression, Somatic Hypermutation, and Memory B Cell Generation in Humoral Immunity Ariel L. -

Molecular Regulation of Peripheral B Cells and Their Progeny in Immunity
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Molecular regulation of peripheral B cells and their progeny in immunity Mark R. Boothby,1,2 Emily Hodges,3 and James W. Thomas1,2 1Department of Pathology–Microbiology–Immunology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232, USA; 2Department of Medicine, Rheumatology Division, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA; 3Department of Biochemistry, Vanderbilt Genetics Institute, Nashville, Tennessee 37232, USA Mature B lymphocytes are crucial components of adap- potentials. A vast trove of findings illuminates the tran- tive immunity, a system essential for the evolutionary fit- scriptional regulation and chromatin modifications (for ness of mammals. Adaptive lymphocyte function requires convenience, referred to here as epigenetic) that program an initially naïve cell to proliferate extensively and its developmental progression from common lymphoid pro- progeny to have the capacity to assume a variety of fates. genitors (CLPs) to the establishment of the naïve popu- These include either terminal differentiation (the long- lations of mature T and B cells (e.g., for review, see lived plasma cell) or metastable transcriptional repro- Busslinger 2004; Champhekar et al. 2015). Similarly, the gramming (germinal center and memory B cells). In this process of diversifying subsets of T cells after their activa- review, we focus principally on the regulation of differen- tion has been studied and reviewed intensively (Glimcher tiation and functional diversification of the “B2” subset. and Murphy 2000; Fang and Zhu 2017; Henning et al. An overview is combined with an account of more recent 2018). Mature B lymphocytes also have the potential to advances, including initial work on mechanisms that distribute their progeny among several distinct fates or eliminate DNA methylation and potential links between intermediate states after they have encountered a ligand intracellular metabolites and chromatin editing. -
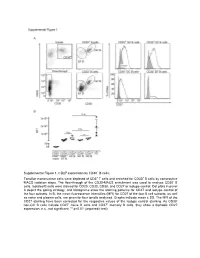
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Molecular Insights Into the Pathogenesis of Follicular Lymphoma
13 Review Article Page 1 of 13 Molecular insights into the pathogenesis of follicular lymphoma Ting Zhou, Stefania Pittaluga, Elaine S. Jaffe Hematopathology Section, Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: None; (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: None; (V) Data analysis and interpretation: None; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Dr. Elaine S. Jaffe. 10 Center Drive, Room 3S 235, National Institutes of Health, Bethesda, MD 20892, USA. Email: [email protected]. Abstract: Follicular lymphoma (FL) represents a group of B-cell neoplasms derived from germinal center (GC) B cells. A better understanding of the pathogenic mechanisms of FL is important for developing innovative therapies. The most common form of FL is primarily nodal and associated with the t(14;18). Recent advances obtained via genomic profiling have provided unprecedented insights into the pathogenesis of FL. Conventional FL evolves through multiple independent or convergent genetic pathways. The classical pathogenesis of t(14;18)-positive FL is a multi-stage and multi-hit process escalating along accumulation of genetic and epigenetic alterations, which starts from FL-like B cells, through premalignant lesions, into full-blown malignancy. Early precursor lesions have been recognized in the form of FL-like B cells in normal peripheral blood, and both in-situ follicular neoplasia (ISFN) and duodenal-type FL. In comparison, t(14;18)-negative FL is much more heterogeneous at the molecular level, and the underlying mechanisms are less well understood. -

B Cells Centrocytes Within Human Germinal Center Discriminates
CXCR4 Expression Functionally Discriminates Centroblasts versus Centrocytes within Human Germinal Center B Cells This information is current as of September 28, 2021. Gersende Caron, Simon Le Gallou, Thierry Lamy, Karin Tarte and Thierry Fest J Immunol 2009; 182:7595-7602; ; doi: 10.4049/jimmunol.0804272 http://www.jimmunol.org/content/182/12/7595 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2009/06/02/182.12.7595.DC1 Material http://www.jimmunol.org/ References This article cites 27 articles, 12 of which you can access for free at: http://www.jimmunol.org/content/182/12/7595.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 28, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology CXCR4 Expression Functionally Discriminates Centroblasts versus Centrocytes within Human Germinal Center B Cells1 Gersende Caron,2*† Simon Le Gallou,2* Thierry Lamy,*‡ Karin Tarte,*† and Thierry Fest3*† The human germinal center is a highly dynamic structure where B cells conduct their terminal differentiation and traffic following chemokine gradients.