Semiflexible Polymers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Information on the Surfaces Not Included in the Basic Content of the Encyclopedia
Brief Information on the Surfaces Not Included in the Basic Content of the Encyclopedia Brief information on some classes of the surfaces which cylinders, cones and ortoid ruled surfaces with a constant were not picked out into the special section in the encyclo- distribution parameter possess this property. Other properties pedia is presented at the part “Surfaces”, where rather known of these surfaces are considered as well. groups of the surfaces are given. It is known, that the Plücker conoid carries two-para- At this section, the less known surfaces are noted. For metrical family of ellipses. The straight lines, perpendicular some reason or other, the authors could not look through to the planes of these ellipses and passing through their some primary sources and that is why these surfaces were centers, form the right congruence which is an algebraic not included in the basic contents of the encyclopedia. In the congruence of the4th order of the 2nd class. This congru- basis contents of the book, the authors did not include the ence attracted attention of D. Palman [8] who studied its surfaces that are very interesting with mathematical point of properties. Taking into account, that on the Plücker conoid, view but having pure cognitive interest and imagined with ∞2 of conic cross-sections are disposed, O. Bottema [9] difficultly in real engineering and architectural structures. examined the congruence of the normals to the planes of Non-orientable surfaces may be represented as kinematics these conic cross-sections passed through their centers and surfaces with ruled or curvilinear generatrixes and may be prescribed a number of the properties of a congruence of given on a picture. -

Linguistic Presentation of Objects
Zygmunt Ryznar dr emeritus Cracow Poland [email protected] Linguistic presentation of objects (types,structures,relations) Abstract Paper presents phrases for object specification in terms of structure, relations and dynamics. An applied notation provides a very concise (less words more contents) description of object. Keywords object relations, role, object type, object dynamics, geometric presentation Introduction This paper is based on OSL (Object Specification Language) [3] dedicated to present the various structures of objects, their relations and dynamics (events, actions and processes). Jay W.Forrester author of fundamental work "Industrial Dynamics”[4] considers the importance of relations: “the structure of interconnections and the interactions are often far more important than the parts of system”. Notation <!...> comment < > container of (phrase,name...) ≡> link to something external (outside of definition)) <def > </def> start-end of definition <spec> </spec> start-end of specification <beg> <end> start-end of section [..] or {..} 1 list of assigned keywords :[ or :{ structure ^<name> optional item (..) list of items xxxx(..) name of list = value assignment @ mark of special attribute,feature,property @dark unknown, to obtain, to discover :: belongs to : equivalent name (e.g.shortname) # number of |name| executive/operational object ppppXxxx name of item Xxxx with prefix ‘pppp ‘ XXXX basic object UUUU.xxxx xxxx object belonged to UUUU object class & / conjunctions ‘and’ ‘or’ 1 If using Latex editor we suggest [..] brackets -

Some Curves and the Lengths of Their Arcs Amelia Carolina Sparavigna
Some Curves and the Lengths of their Arcs Amelia Carolina Sparavigna To cite this version: Amelia Carolina Sparavigna. Some Curves and the Lengths of their Arcs. 2021. hal-03236909 HAL Id: hal-03236909 https://hal.archives-ouvertes.fr/hal-03236909 Preprint submitted on 26 May 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Some Curves and the Lengths of their Arcs Amelia Carolina Sparavigna Department of Applied Science and Technology Politecnico di Torino Here we consider some problems from the Finkel's solution book, concerning the length of curves. The curves are Cissoid of Diocles, Conchoid of Nicomedes, Lemniscate of Bernoulli, Versiera of Agnesi, Limaçon, Quadratrix, Spiral of Archimedes, Reciprocal or Hyperbolic spiral, the Lituus, Logarithmic spiral, Curve of Pursuit, a curve on the cone and the Loxodrome. The Versiera will be discussed in detail and the link of its name to the Versine function. Torino, 2 May 2021, DOI: 10.5281/zenodo.4732881 Here we consider some of the problems propose in the Finkel's solution book, having the full title: A mathematical solution book containing systematic solutions of many of the most difficult problems, Taken from the Leading Authors on Arithmetic and Algebra, Many Problems and Solutions from Geometry, Trigonometry and Calculus, Many Problems and Solutions from the Leading Mathematical Journals of the United States, and Many Original Problems and Solutions. -

Spiral Pdf, Epub, Ebook
SPIRAL PDF, EPUB, EBOOK Roderick Gordon,Brian Williams | 496 pages | 01 Sep 2011 | Chicken House Ltd | 9781906427849 | English | Somerset, United Kingdom Spiral PDF Book Clear your history. Cann is on the run. Remark: a rhumb line is not a spherical spiral in this sense. The spiral has inspired artists throughout the ages. Metacritic Reviews. Fast, Simple and effective in getting high quality formative assessment in seconds. It has been nominated at the Globes de Cristal Awards four times, winning once. Name that government! This last season has two episodes less than the previous ones. The loxodrome has an infinite number of revolutions , with the separation between them decreasing as the curve approaches either of the poles, unlike an Archimedean spiral which maintains uniform line-spacing regardless of radius. Ali Tewfik Jellab changes from recurring character to main. William Schenk Christopher Tai Looking for a movie the entire family can enjoy? Looking for a movie the entire family can enjoy? Edit Cast Series cast summary: Caroline Proust June Assess in real-time or asynchronously. Time Traveler for spiral The first known use of spiral was in See more words from the same year. A hyperbolic spiral appears as image of a helix with a special central projection see diagram. TV series to watch. External Sites. That dark, messy, morally ambivalent universe they live in is recognisable even past the cultural differences, such as the astonishing blurring of the boundary between investigative police work and judgement — it's not so much uniquely French as uniquely modern. Photo Gallery. Some familiar faces, and some new characters, keep things ticking along nicely. -

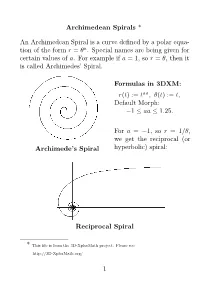
Archimedean Spirals ∗
Archimedean Spirals ∗ An Archimedean Spiral is a curve defined by a polar equation of the form r = θa, with special names being given for certain values of a. For example if a = 1, so r = θ, then it is called Archimedes’ Spiral. Archimede’s Spiral For a = −1, so r = 1/θ, we get the reciprocal (or hyperbolic) spiral. Reciprocal Spiral ∗This file is from the 3D-XploreMath project. You can find it on the web by searching the name. 1 √ The case a = 1/2, so r = θ, is called the Fermat (or hyperbolic) spiral. Fermat’s Spiral √ While a = −1/2, or r = 1/ θ, it is called the Lituus. Lituus In 3D-XplorMath, you can change the parameter a by going to the menu Settings → Set Parameters, and change the value of aa. You can see an animation of Archimedean spirals where the exponent a varies gradually, from the menu Animate → Morph. 2 The reason that the parabolic spiral and the hyperbolic spiral are so named is that their equations in polar coordinates, rθ = 1 and r2 = θ, respectively resembles the equations for a hyperbola (xy = 1) and parabola (x2 = y) in rectangular coordinates. The hyperbolic spiral is also called reciprocal spiral because it is the inverse curve of Archimedes’ spiral, with inversion center at the origin. The inversion curve of any Archimedean spirals with respect to a circle as center is another Archimedean spiral, scaled by the square of the radius of the circle. This is easily seen as follows. If a point P in the plane has polar coordinates (r, θ), then under inversion in the circle of radius b centered at the origin, it gets mapped to the point P 0 with polar coordinates (b2/r, θ), so that points having polar coordinates (ta, θ) are mapped to points having polar coordinates (b2t−a, θ). -

Analysis of Spiral Curves in Traditional Cultures
Forum _______________________________________________________________ Forma, 22, 133–139, 2007 Analysis of Spiral Curves in Traditional Cultures Ryuji TAKAKI1 and Nobutaka UEDA2 1Kobe Design University, Nishi-ku, Kobe, Hyogo 651-2196, Japan 2Hiroshima-Gakuin, Nishi-ku, Hiroshima, Hiroshima 733-0875, Japan *E-mail address: [email protected] *E-mail address: [email protected] (Received November 3, 2006; Accepted August 10, 2007) Keywords: Spiral, Curvature, Logarithmic Spiral, Archimedean Spiral, Vortex Abstract. A method is proposed to characterize and classify shapes of plane spirals, and it is applied to some spiral patterns from historical monuments and vortices observed in an experiment. The method is based on a relation between the length variable along a curve and the radius of its local curvature. Examples treated in this paper seem to be classified into four types, i.e. those of the Archimedean spiral, the logarithmic spiral, the elliptic vortex and the hyperbolic spiral. 1. Introduction Spiral patterns are seen in many cultures in the world from ancient ages. They give us a strong impression and remind us of energy of the nature. Human beings have been familiar to natural phenomena and natural objects with spiral motions or spiral shapes, such as swirling water flows, swirling winds, winding stems of vines and winding snakes. It is easy to imagine that powerfulness of these phenomena and objects gave people a motivation to design spiral shapes in monuments, patterns of cloths and crafts after spiral shapes observed in their daily lives. Therefore, it would be reasonable to expect that spiral patterns in different cultures have the same geometrical properties, or at least are classified into a small number of common types. -

Mathematics of the Extratropical Cyclone Vortex in the Southern
ACCESS Freely available online y & W OPEN log ea to th a e im r l F C o f r e o c l Journal of a a s n t r i n u g o J ISSN: 2332-2594 Climatology & Weather Forecasting Case Report Mathematics of the Extra-Tropical Cyclone Vortex in the Southern Atlantic Ocean Gobato R1*, Heidari A2, Mitra A3 1Green Land Landscaping and Gardening, Seedling Growth Laboratory, 86130-000, Parana, Brazil; 2Aculty of Chemistry, California South University, 14731 Comet St. Irvine, CA 92604, USA; 3Department of Marine Science, University of Calcutta, 35 B.C. Road Kolkata, 700019, India ABSTRACT The characteristic shape of hurricanes, cyclones, typhoons is a spiral. There are several types of turns, and determining the characteristic equation of which spiral CB fits into is the goal of the work. In mathematics, a spiral is a curve which emanates from a point, moving farther away as it revolves around the point. An “explosive extra-tropical cyclone” is an atmospheric phenomenon that occurs when there is a very rapid drop in central atmospheric pressure. This phenomenon, with its characteristic of rapidly lowering the pressure in its interior, generates very intense winds and for this reason it is called explosive cyclone, bomb cyclone. It was determined the mathematical equation of the shape of the extratropical cyclone, being in the shape of a spiral called "Cotes' Spiral". In the case of Cyclone Bomb (CB), which formed in the south of the Atlantic Ocean, and passed through the south coast of Brazil in July 2020, causing great damages in several cities in the State of Santa Catarina. -
Archimedean Spirals * an Archimedean Spiral Is a Curve
Archimedean Spirals * An Archimedean Spiral is a curve defined by a polar equa- tion of the form r = θa. Special names are being given for certain values of a. For example if a = 1, so r = θ, then it is called Archimedes’ Spiral. Formulas in 3DXM: r(t) := taa, θ(t) := t, Default Morph: 1 aa 1.25. − ≤ ≤ For a = 1, so r = 1/θ, we get the− reciprocal (or Archimede’s Spiral hyperbolic) spiral: Reciprocal Spiral * This file is from the 3D-XplorMath project. Please see: http://3D-XplorMath.org/ 1 The case a = 1/2, so r = √θ, is called the Fermat (or hyperbolic) spiral. Fermat’s Spiral While a = 1/2, or r = 1/√θ, it is called the Lituus: − Lituus 2 In 3D-XplorMath, you can change the parameter a by go- ing to the menu Settings Set Parameters, and change the value of aa. You can see→an animation of Archimedean spirals where the exponent a = aa varies gradually, be- tween 1 and 1.25. See the Animate Menu, entry Morph. − The reason that the parabolic spiral and the hyperbolic spiral are so named is that their equations in polar coor- dinates, rθ = 1 and r2 = θ, respectively resembles the equations for a hyperbola (xy = 1) and parabola (x2 = y) in rectangular coordinates. The hyperbolic spiral is also called reciprocal spiral be- cause it is the inverse curve of Archimedes’ spiral, with inversion center at the origin. The inversion curve of any Archimedean spirals with re- spect to a circle as center is another Archimedean spiral, scaled by the square of the radius of the circle. -

Chemotaxis of Sperm Cells
Chemotaxis of sperm cells Benjamin M. Friedrich* and Frank Ju¨licher* Max Planck Institute for the Physics of Complex Systems, No¨thnitzer Strasse 38, 01187 Dresden, Germany Edited by Charles S. Peskin, New York University, New York, NY, and approved June 20, 2007 (received for review April 17, 2007) We develop a theoretical description of sperm chemotaxis. Sperm In this case, sperm swim on helical paths. In the presence of a cells of many species are guided to the egg by chemoattractants, chemoattractant concentration gradient, the helices bend, even- a process called chemotaxis. Motor proteins in the flagellum of the tually leading to alignment of the helix axis with the gradient (8). sperm generate a regular beat of the flagellum, which propels the Chemotaxis is mediated by a signaling system that is located sperm in a fluid. In the absence of a chemoattractant, sperm swim in the sperm flagellum (10). Specific receptors in the flagellar in circles in two dimensions and along helical paths in three membrane are activated upon binding of chemoattractant mol- dimensions. Chemoattractants stimulate a signaling system in the ecules and start the production of cyclic guanine monophosphate flagellum, which regulates the motors to control sperm swimming. (cGMP). A rise in cGMP gates the opening of potassium Our theoretical description of sperm chemotaxis in two and three channels and causes a hyperpolarization of the flagellar mem- dimensions is based on a generic signaling module that regulates brane. This hyperpolarization triggers the opening of voltage- the curvature and torsion of the swimming path. In the presence gated calcium channels and the membrane depolarizes again. -

OSL - Object Specification Language
Zygmunt Ryznar dr emeritus Cracow Poland [email protected] OSL - Object Specification Language (includes BSL,SSL,HSL,geometric structures & brain definition) Abstract OSL© is a markup descriptive language for a simple formal description of any object in terms of structure and behavior. In this paper we present a geometric approach, the kernel and subsets for IT system, business and human-being as a proof of universality. This language was invented as a tool for a free structured modeling and design. Keywords free structured modeling and design, OSL, object specification language, human descriptive language, factual markup, geometric view, object specification, GSO -geometrically structured objects. I. INTRODUCTION Generally, a free structured modeling is a collection of methods, techniques and tools for modeling the variable structure as an opposite to the widely used “well-structured” approach focused on top-down hierarchical decomposition. It is assumed that system boundaries are not finally defined, because the system is never to be completed and components are are ready to be modified at any time. OSL is dedicated to present the various structures of objects, their relations and dynamics (events, actions and processes). This language may be counted among markup languages. The fundamentals of markup languages were created in SGML (Standard Generalized Markup Language) [6] descended from GML (name comes from first letters of surnames of authors: Goldfarb, Mosher, Lorie, later renamed as Generalized Markup Language) developed at IBM [15]. Markups are usually focused on: - documents (tags: title, abstract, keywords, menu, footnote etc.) e.g. latex [14], - objects (e.g. OSL), - internet pages (tags used in html, xml – based on SGML [8] rules), - data (e.g. -

On the Relevance of the Differential Expressions $ F^ 2+ F'^ 2$, $ F+ F
On the relevance of the differential expressions 2 2 2 f + f ′ , f + f ′′ and ff ′′ f ′ for the geometrical and mechanical− properties of curves James Bell Cooper Contents 1 Introduction 4 2 The Kepler problem 9 2.1 Kepler’ssecondlaw........................ 9 2.2 Theinversesquarelaw ...................... 10 2.3 The Newton-Somerville equation . 11 2.4 Acriterionforapowerlaw. 13 2.5 Someapplications......................... 13 2.6 Twosimplecases ......................... 14 3 The Affine Curvature of an Orbit Determines the Force Law 16 3.1 Further geometric quantities associated with orbits . 16 3.2 Affinecurvature.......................... 17 3.3 Acomputationalproof . 18 arXiv:1102.1579v1 [math-ph] 8 Feb 2011 3.4 Aconceptualproof ........................ 19 4 The Kasner-Arnold duality 20 4.1 Sinusoidal spirals and the d-transformation . 20 4.2 Thegeneralsituation . .. .. 21 5 A Duality between Trajectories for Central and Parallel Force Laws 23 5.1 Curves of the form (F (t), f(t)).................. 24 5.2 The duality (quadrality) . 25 1 5.3 Examplesofduality........................ 26 5.4 The sinusoidal spirals and catenaries . 28 6 On the expressions f 2 + f ′2, f + f ′′, ff ′′ f 2, how to solve the ′ ′′ −′′ equations f 2 + f 2 = af α, f + f = bf β, ff f 2 = cf γ and why one might want to − 29 6.1 The relationship between the equations . 30 6.2 Somesolutions .......................... 30 6.3 Wheretheexpressionsarise . 32 6.4 The calculus of variations . 33 6.5 Twomoresubtleexamples . 34 6.6 Miscellanea ............................ 35 6.6.1 The explicit form of the parametrisation (Fd(t), fd(t)) 35 6.6.2 Theconnectionwithcontactgeometry . 35 6.6.3 Curveswithprescribedcurvature . -

Symmetrical Laws of Structure of Helicoidally-Like Biopolymers in the Framework of Algebraic Topology
Symmetrical laws of structure of helicoidally-like biopolymers in the framework of algebraic topology. III. Nature of the double and relations between the alpha helix and the various forms of DNA structures M.I.Samoylovich, A.L.Talis Central Research and Technology Institute "Technomash", Moscow E-mail: [email protected] Institute of Organoelement Compounds of Russian Academy of Sciences, Moscow Crystallography Reports, in press In the frameworks of algebraic topology α-helix and different DNA-conformations are determined as the local latticed packing, confined by peculiar minimal surfaces which are similar to helicoids. These structures are defined by Weierstrass representation and satisfy to a zero condition for the instability index of the surface and availability of bifurcation points for these surfaces. The topological stability of such structures corresponds to removing of the configuration degeneracy and to existence of bifurcation points for these surfaces. The considered ordered non-crystalline structures are determined by homogeneous manifolds - algebraic polytopes, corresponding to the definite substructures the 8-dimensional lattice E8.The joining of two semi-turns of two spirals into the turn of a single two-spiral (helical) system is effected by the topological operation of a connected sum. The applied apparatus permits to determine a priori the symmetry parameters of the double spirals in A, B and Z forms DNA structures. 1. Introduction .Properties of minimal surfaces and their presetting by the Weierstrass representation One difficulty of local description of non-crystalline systems consists in the necessity to consider their ordering automorphisms, playing the role (as a rule, in a parametric way) of customary symmetry elements; therefore, in present work, a special attention will be given to visualization of algebraic elements and one-parameter subgroups used to achieve this.