Phase 2 Randomized, Placebo-Controlled
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Multicenter, Multi-Outbreak, Randomized, Controlled Safety And
2018 Ebola MCM RCT Protocol Page 1 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 A Multicenter, Multi-Outbreak, Randomized, Controlled Safety and Efficacy Study of Investigational Therapeutics for the Treatment of Patients with Ebola Virus Disease Short Title: 2018 Ebola MCM RCT Protocol Sponsored by: Office of Clinical Research Policy and Regulatory Operations (OCRPRO) National Institute of Allergy and Infectious Diseases 5601 Fishers Lane Bethesda, MD 20892 NIH Protocol Number: 19-I-0003 Protocol IND #: 125530 ClinicalTrials.gov Number: NCT03719586 Date: 4 October 2019 Version: 7.0 CONFIDENTIAL This document is confidential. No part of it may be transmitted, reproduced, published, or used by other persons without prior written authorization from the study sponsor and principal investigator. 2018 Ebola MCM RCT Protocol Page 2 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 KEY ROLES DRC Principal Investigator: Jean-Jacques Muyembe-Tamfum, MD, PhD Director-General, DRC National Institute for Biomedical Research Professor of Microbiology, Kinshasa University Medical School Kinshasa Gombe Democratic Republic of the Congo Phone: +243 898949289 Email: [email protected] Other International Investigators: see Appendix E Statistical Lead: Lori Dodd, PhD Biostatistics Research Branch, DCR, NIAID 5601 Fishers Lane, Room 4C31 Rockville, MD 20852 Phone: 240-669-5247 Email: [email protected] U.S. Principal Investigator: Richard T. Davey, Jr., MD Clinical Research Section, LIR, NIAID, NIH Building 10, Room 4-1479, Bethesda, -

Recent Zoonotic and Vector-Borne Viral Threats NIAID Vaccine
4/1/2020 PROTOTYPE PATHOGEN APPROACH TO PANDEMIC PREPAREDNESS HIV→PNEUMOVIRUS→PARAMYXOVIRUS→CORONAVIRUS ADVAC Alumni 2 April 2020 Barney S. Graham, MD, PhD Deputy Director Vaccine Research Center, NIAID, NIH 1 Recent Zoonotic and Vector-borne Viral Threats • Hanta virus • Nipah/Hendra • West Nile virus • SARS • Influenza • Chikungunya • Ebola • MERS • Zika • EV‐D68 • SARS‐CoV‐2 2 NIAID Vaccine Research Center Basic Research Process Development Nucleic acid Vectors VLPs • AIDS/HIV • Influenza Proteins and cGMP Manufacturing nanoparticles • Ebola/Marburg • RSV • Malaria Monoclonal antibodies • Tuberculosis • EID GLP Analysis • West Nile virus, Zika • Chikungunya • W/E/V equine encephalitis viruses • MERS‐CoV, SARS, and other CoV • Nipah and other paramyxoviruses • EV‐D68 and other picornaviruses Clinical Trials • Smallpox 3 1 4/1/2020 Public health burden of re-emerging & emerging viruses Traditional Approaches • Licensed vaccines/antibiotics Vaccine • Passive surveillance Challenges • Contact tracing • Quarantine • Vaccines for unmet needs • Emerging viruses • Improving licensed vaccines 4 4 New Technologies are Transforming Vaccinology • Structure-based vaccine design Structural analysis of antigenic sites on viral • Single-cell sorting, sequencing, and bioinformatics surface glycoproteins – Rapid isolation of human mAbs Isolation of human monoclonal – Definition of antibody lineages antibodies from single B cells – Analysis of immune responses • Protein engineering of self-assembling nanoparticles • Rapid DNA synthesis • Recombinant -

Kizzmekia S. Corbett
KIZZMEKIA S. CORBETT, PHD Senior Research Fellow National Institutes of Health | National Institute of Allergy and Infectious Diseases | Vaccine Research Center 40 Convent Drive, Building 40 - Room 2608, Bethesda MD 20892 [email protected] | phone: 301.761.7610 | fax: 301.480.2771 EDUCATION Doctor of Philosophy in Microbiology and Immunology 2014 University of North Carolina at Chapel Hill (UNC) Director’s Scholar Bachelor of Science in Biological Sciences | Secondary Major in Sociology 2008 University of Maryland – Baltimore County (UMBC) Robert and Jane Meyerhoff Scholar RESEARCH EXPERIENCE Research Fellow 10/14 - present National Institutes of Health | Vaccine Research Center | PI: Barney S. Graham, MD PhD Research Interest: mechanisms of viral immunity to inform influenza and coronavirus vaccine development Graduate Research Assistant 08/09 – 10/14 University of North Carolina at Chapel Hill | Microbiology and Immunology | PI: Aravinda de Silva, MPH PhD Research Interest: human antibody responses to dengue virus infection Visiting Scholar 04/14 – 05/14 Genetech Research Institute | PI: Aruna Dharshan de Silva, PhD Research Interest: immunological relevance of dengue virion maturation Postbaccalaureate Research Fellow 05/08 – 08/09 National Institutes of Health | Vaccine Research Center | PI: Barney S. Graham, MD PhD Research Interest: novel vaccine platform design for respiratory syncytial virus antigens NIH Undergraduate Scholarship Program Summer Intern 06/07 – 08/07 National Institutes of Health | Vaccine Research Center -

Scientific Consultation on Zika Virus Vaccine Development
World Health Organization and National Institute of Allergy and Infectious Diseases, National Institutes of Health Scientific Consultation on Zika Virus Vaccine Development January 10–11, 2017 5601 Fishers Lane Rockville, Maryland Meeting Summary Tuesday, January 10, 2017 INTRODUCTION Anthony S. Fauci, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), United States The National Institute of Allergy and Infectious Diseases (NIAID) and the World Health Organization (WHO) convened the Scientific Consultation on Zika Virus Vaccine Development to discuss challenges and recent advances in the development of Zika virus (ZIKV) vaccines. The NIAID Director opened the meeting by stating that the recent spread of ZIKV mirrors that of other emerging arboviruses in the Americas in the past several years, including chikungunya, West Nile, and dengue. The NIAID research response to Zika builds on those experiences. A primary goal of this research is to develop medical countermeasures including diagnostics, therapeutics, and vaccines. Challenges specific to the development of a Zika vaccine include: the lack of adequate animal models; uncertainties in the epidemiology, which affect clinical trial site selection; the likelihood that the vaccine will need to induce sterilizing immunity to prevent congenital Zika syndrome (CZS); preexisting immunity to other flaviviruses in some regions; and potential specific risks such as Guillain-Barré syndrome (GBS) and the administration of live vaccines to pregnant women. Several Zika vaccine candidates are being developed with different development timelines and likely target populations. The planned timelines include large, well-controlled clinical trials to evaluate safety and efficacy, and if successful, subsequent licensure; should public health need and available safety and efficacy data justify deployment of vaccine before licensure, access through appropriate regulatory mechanisms could be considered. -

Vaccine Research Center, NIAID, NIH
4/17/2018 PIPELINE OF HIV MONOCLONAL ANTIBODIES FOR PREVENTION OF HIV Session on Antibody‐Mediated Prevention 2018 Global Vaccine and Immunization Research Forum (GVIRF) 20‐22 March 2018 Bangkok, Thailand Barney S. Graham, MD, PhD Deputy Director Vaccine Research Center, NIAID, NIH Dale and Betty Bumpers Vaccine Research Center Vaccine Research Center National Institute of Allergy and Infectious Diseases National Institutes of Health 2017 VRC Principal Investigators and Program Directors 1 4/17/2018 VRC Research & Development: From AIDS to Zika August 2000 AIDS/HIV Chikungunya Ebola/Marburg Influenza Malaria MERS-CoV, SARS RSV Smallpox Tuberculosis W/E/V equine encephalitis viruses West Nile virus, Zika Engineering Lab Pilot Plant Vaccine Evaluation Clinic Immunology Lab Partners for Advanced Development Functional Activity of Anti-Viral Antibodies • Neutralization – Aggregation – Attachment blocking – Cleavage inhibition – Fusion inhibition – Preventing particle release • Fc‐mediated functions – Antibody dependent cell‐mediated cytotoxicity (NK, Macrophage, Neutrophil) – Complement binding and activation • Opsinization and clearance by non‐susceptible cells • Blocking pathogenic immunomodulatory molecules 2 4/17/2018 Long History of Using Antibodies to Treat Infectious Diseases (Serum Therapy) Shibasaburo Kitasato 1890: Emil von Behring and Shibasaburo Kitasato worked on “anti‐ toxins” for tetanus and diphtheria that led to concept for serum therapy 1901: Emil von Behring ‐ 1901 Nobel Prize in Physiology or Medicine “For his work on serum therapy, especially its application against diphtheria, by which he has opened a new road in the domain of Wernicke, Frosch, and medical science and thereby placed in the hands of the physician a Behring in Koch’s Berlin Lab victorious weapon against illness and deaths". -

Identification and Use of Biomarkers to Advance Development of Preventive Vaccines
FDA/NIH/CEPI BIOMARKERS WORKSHOP Identification and Use of Biomarkers to Advance Development of Preventive Vaccines September 16 & 17, 2019 5601 Fishers Lane, Rm. 1D13 Rockville, MD 20852 Meeting goals: Recent legislation, including the 21st Century Cures Act and Prescription Drug User Fee Act (PDUFA) VI, encourages use of biomarkers to enhance development and approval of new and innovative drug and biological products. Particularly in the field of vaccines to prevent infectious disease, a well-characterized biomarker has tremendous potential value, because it can enhance basic research, facilitate vaccine development, and guide the effective use of vaccines. To facilitate the realization of this potential, FDA’s Center for Biologics Evaluation and Research (CBER), NIH’s National Institute for Allergy and Infectious Diseases (NIAID), and the Coalition for Epidemic Preparedness Innovations (CEPI) are partnering to convene this workshop. The purpose is to exchange information with stakeholders from industry, academia, and government about the scientific, clinical, and regulatory challenges encountered in the discovery, characterization, and qualification of biomarkers for preventive vaccines for infectious diseases indications. The objectives of the workshop include: • Provide the context and understand the importance of biomarkers in vaccine discovery and development, including through review of successful case examples. • Clarify the regulatory framework that informs the use of biomarkers in vaccine development and licensure. • Assess the quality of the evidence for biomarkers to support decisions (regulatory, programmatic, and otherwise) regarding candidate and licensed vaccine products for specific infectious diseases. • Explore how new technologies and innovations can be applied to advance the science of vaccine-associated biomarkers. • Understand the institutional perspectives/priorities with respect to the use of biomarkers for vaccine development and deployment across a wide range of stakeholders. -

Official Title: a Phase 1 Open Label, Dose-Escalation Clinical Trial To
Official Title: A Phase 1 Open Label, Dose-Escalation Clinical Trial to Evaluate the Safety and Immunogenicity of a Trivalent Virus-Like Particle (VLP) Encephalitis Vaccine, VRC-WEVVLP073-00-VP, in Healthy Adults ClinicalTrials.gov Identifier: NCT03879603 Document Type: • Redacted Study Protocol, including Letters of Amendment (Statistical Analysis Considerations located in Section 6 of the Protocol) • Redacted IRB-approved Version 2.0 Informed Consent (dated March 12, 2019) Document/Date: • Protocol Version 1.0: January 9, 2019 • Protocol Version 1.0, Letter of Amendment #1: March 12, 2019 • Protocol Version 1.0, Letter of Amendment #2: April 23, 2019 • Protocol Version 1.0, Letter of Amendment #3: October 24, 2019 • Version 2.0 IRB-approved Informed Consent (dated March 12, 2019 with March 18, 2019 IRB- approval) Version 1.0 January 9, 2019 VACCINE RESEARCH CENTER Protocol VRC 313 A PHASE 1 OPEN LABEL, DOSE-ESCALATION CLINICAL TRIAL TO EVALUATE THE SAFETY AND IMMUNOGENICITY OF A TRIVALENT VIRUS-LIKE PARTICLE (VLP) ENCEPHALITIS VACCINE, VRC-WEVVLP073-00-VP, IN HEALTHY ADULTS Vaccine Provided by National Institute of Allergy and Infectious Diseases (NIAID) Vaccine Research Center (VRC) Bethesda, Maryland Clinical Trial Sponsored by National Institute of Allergy and Infectious Diseases (NIAID) Vaccine Research Center (VRC) Bethesda, Maryland BB-IND 11738 – held by VRC, NIAID, NIH Clinical Trial Management Leidos Biomedical Research, Inc. Frederick, Maryland, USA Protocol Chairs Julie Ledgerwood, D.O. Cristina A. Carter, M.D. VRC, NIAID, NIH Bethesda, MD 20892 Clinical Research Site Principal Investigator: Srilatha Edupuganti, MD; MPH IRB Initial Review Date: February 20, 2019 Confidentiality Statement This document is confidential and is to be distributed for review only to investigators, potential investigators, consultants, study staff, and applicable independent ethics committees or institutional review boards. -

Harmonization of Zika Neutralization Assays by Using the WHO International Standard for Anti-Zika Virus Antibody
www.nature.com/npjvaccines ARTICLE OPEN Harmonization of Zika neutralization assays by using the WHO International Standard for anti-Zika virus antibody Giada Mattiuzzo 1*, Ivana Knezevic 2, Mark Hassall1, James Ashall1, Sophie Myhill1, Valwynne Faulkner1, Jason Hockley3, Peter Rigsby 3, Dianna E. Wilkinson1, Mark Page 1 and the collaborative study participants During outbreaks of emerging viruses, such as the Zika outbreak in 2015–2016, speed and accuracy in detection of infection are critical factors to control the spread of the disease; often serological and diagnostic methods for emerging viruses are not well developed and validated. Thus, vaccines and treatments are difficult to evaluate due to the lack of comparable methods. In this study, we show how the 1st WHO International Standard for anti-Zika antibody was able to harmonize the neutralization titres of a panel of serological Zika-positive samples from laboratories worldwide. Expression of the titres in International Unit per millilitre reduced the inter-laboratory variance, allowing for greater comparability between laboratories. We advocate the use of the International Standard for anti-Zika virus antibodies for the calibration of neutralization assays to create a common language, which will permit a clear evaluation of the results of different clinical trials and expedite the vaccine/treatment development. npj Vaccines (2019) ; https://doi.org/10.1038/s41541-019-0135-34:42 1234567890():,; INTRODUCTION development of these products is ongoing. According to the Zika virus (ZIKV) is a flavivirus mainly transmitted by Aedes species current knowledge on the transmission of ZIKV and experiences mosquitoes. ZIKV was discovered in 1947 and was only known as a with past disease outbreaks, WHO has prioritized the development cause of sporadic mild disease in Africa and Asia.1,2 The first major of vaccines suitable for use in an emergency or outbreak scenario. -

Curriculum Vitae
KIZZMEKIA S. CORBETT, PHD Senior Research Fellow National Institutes of Health | National Institute of Allergy and Infectious Diseases | Vaccine Research Center 40 Convent Drive, Building 40 - Room 2608, Bethesda MD 20892 [email protected] | personal: 919.218.9101 | lab: 301.761.7610 | fax: 301.480.2771 EDUCATION Doctor of Philosophy in Microbiology and Immunology 2014 University of North Carolina at Chapel Hill (UNC) Director’s Scholar Bachelor of Science in Biological Sciences | Secondary Major in Sociology 2008 University of Maryland – Baltimore County (UMBC) Robert and Jane Meyerhoff Scholar RESEARCH EXPERIENCE Research Fellow 10/14 - present National Institutes of Health | Vaccine Research Center | PI: Barney S. Graham, MD PhD Research Interest: mechanisms of viral immunity to inform influenza and coronavirus vaccine development Graduate Research Assistant 08/09 – 10/14 University of North Carolina at Chapel Hill | Microbiology and Immunology | PI: Aravinda de Silva, MPH PhD Research Interest: human antibody responses to dengue virus infection Visiting Scholar 04/14 – 05/14 Genetech Research Institute | PI: Aruna Dharshan de Silva, PhD Research Interest: immunological relevance of dengue virion maturation Postbaccalaureate Research Fellow 05/08 – 08/09 National Institutes of Health | Vaccine Research Center | PI: Barney S. Graham, MD PhD Research Interest: novel vaccine platform design for respiratory syncytial virus antigens NIH Undergraduate Scholarship Program Summer Intern 06/07 – 08/07 National Institutes of Health | -

Meeting Transcript
This transcript appears as received from the commercial transcribing service after inclusion of minor corrections to typographical and factual errors recommended by the Technical Point of Contact. 1 2 3 4 5 6 7 ADVANCING SCIENTIFIC AND REGULATORY APPROACHES 8 FOR USE AND DEVELOPMENT OF BIOMARKERS 9 FOR PREVENTATIVE VACCINES 10 MEETING 11 MONDAY, SEPTEMBER 16, 2019 12 8:34 A.M. 13 5601 FISHERS LANE 14 ROOM 1D13 15 ROCKVILLE, MARYLAND 20852 16 17 18 19 20 21 22 23 24 25 1 APPEARANCES 2 SPEAKERS: 3 MARION GRUBER, PHD - DIRECTOR, 4 OFFICE OF VACCINES RESEARCH AND REVIEW CBER/FDA 5 6 DEBRA YESKEY, PHARMD - HEAD OF REGULATORY AFFAIRS, 7 NORTH AMERICA CEPI 8 9 JEFF ROBERTS, MD - ASSOCIATE DIRECTOR FOR MEDICAL 10 COUNTERMEASURES AND SCIENTIFIC AFFAIRS OVRR/CBER/FDA 11 12 BARNEY GRAHAM, MD, PHD - DEPUTY DIRECTOR, 13 VACCINE RESEARCH CENTER, CHIEF 14 VIRAL PATHOGENESIS LABORATORY AND TRANSLATIONAL 15 SCIENCE CORE VIRAL PATHOGENESIS LABORATORY 16 17 THEODORE PIERSON, PHD - CHIEF, 18 VIRAL PATHOGENESIS SECTION CHIEF, 19 LABORATORY OF VIRAL DISEASES 20 LABORATORY OF VIRAL DISEASE, NIAID 21 22 JULIA LEDGERWOOD, D.O. - CHIEF MEDICAL OFFICER AND CHIEF, 23 CLINICAL TRIALS PROGRAM VACCINE RESEARCH CENTER NIAID, NIH 1 KATRIN RAMSAUER, PHD - CHIEF SCIENTIFIC OFFICER, 2 THEMIS BIOSCIENCE GMBH 3 4 LONG WANG, MD, PHD - DIRECTOR, 5 GLOBAL REGULATORY TEAM LEADER, 6 VACCINES &INFECTIOUS DISEASE MERCK INC. 7 8 NANCY SULLIVAN, PHD - CHIEF, 9 BIODEFENSE RESEARCH SECTION VIRAL PATHOGENESIS LABORATORY, NIAID 10 11 JENNY HENDRICKS, PHD - HEAD BIOMARKERS, 12 VIRAL VACCINES, JANSSEN VACCINES ID&V 13 14 RONG XU, MD, PHD - DIRECTOR OF IMMUNOLOGY, 15 PROFECTUS BIOSCIENCES, INC. -
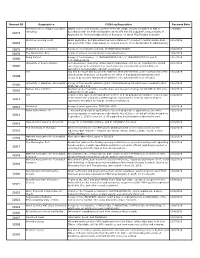
Request ID Organization FOIA Log Description Received Date Stanford John S
Request ID Organization FOIA Log Description Received Date Stanford John S. Knight Journalism Digital copies of all financial conflict of interest (FCOI) reports submitted to NIH in 1/3/2020 51073 fellowship accordance with the Federal regulation at 42 CFR Part 50 Subpart F, Responsibility of Applicants for Promoting Objectivity in Research for which PHS Funding is Sought Skid Row Housing Trust Grant application and associated summary statement for project 1P50MH115846-01A1 , 10/2/2019 52818 sub-project ID 7132. (Date Range for Record Search: From 05/18/2018 To 05/10/2019) 52875 Washington state university A copy of the following contract, HHSN271201100027C. 10/2/2019 52876 The Washington Post Copies of various correspondence and attachments. 10/8/2019 Being Patient A copy of the following: 1) 1R43AG059520-01A1, 2) 1R43AG058357-01 and 3) 10/1/2019 52906 4R44AG055319-02. University of South Carolina Correspondence (including emails and meeting/phone call notes) regarding the animal 10/1/2019 52907 care program at the University of South Carolina, Columbia SC and the Office of Laboratory Animal Welfare (OLAW) employees self Any and all report and documents related to NIH wide internal control assessment of the 10/7/2019 52908 clinical center pharmacy conducted by the office of management assessment and executed by a senior management analyst at the national institutes of health. University of Alabama - Birmingham A copy of the records related to grant #1U01CA224151-01 which were released in NCI 10/2/2019 52909 FOIA Case #19-114. Nathan Kline Institute Abstract, project narrative, specific aims, and research strategy for R01MH101307, plus 10/3/2019 52910 budget info for all years. -

Ebola Virus Vaccine Development
Dale and Betty Bumpers Vaccine Research Center National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services Ebola Virus Vaccine Development Nancy J. Sullivan, Ph.D. Chief, Biodefense Research Section, VRC, NIAID, NIH Les Pensieres, Fondation Merieux Conference Center January 12, 2015 Early Experimental Approaches to Vaccines Against Ebolavirus Killed virus (MBG) vaccine Enhanced disease in guinea pigs Recombinant protein (insect cells) Partial protection in guinea pigs (MBG) No protection in primates Have filoviruses evolved mechanisms to evade humoral immunity? Challenges for Protective Humoral Immunity Against Ebolavirus Pleomorphic (length 14,000nm) and high density glycoprotein…accessible? Stable virion, lethal infection with few particles Broad tropism and multi-organ distribution Rapid internalization via macropinocytosis and use of intracellular receptor Vaccine-Induced Immune Protection Against Ebolavirus Infection DNA/rAd EBOV GP Weeks 0 4 8 20 32 VP30 VP24 L VP40 GP 5’ NP VP35 3’ rAd EBOV IR Overlap Trailer Leader IR Overlap• IRDNA/rAd5Edit Overlap demonstrates proof of principle for vaccine Site protection of macaquesWeeks 0 4 • Single shot rAd5 vaccine provides rapid protection Plasmid DNA Replication-defective rAd 100 NHP: Cynomolgus macaques BSL4, n=4 per group 50 EBOV: IM challenge % Survival 1000 PFU Mortality at 6-9 days 0 No vaccine DNA/rAd5 rAd5 (n>50*) (n=8) (n=23*) Sullivan et al., Nature 2000, 2003 Pathway to Licensure for Ebola and Marburg Vaccines