This Electronic Thesis Or Dissertation Has Been Downloaded from the King’S Research Portal At
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Nuclear PTEN Safeguards Pre-Mrna Splicing to Link Golgi Apparatus for Its Tumor Suppressive Role
ARTICLE DOI: 10.1038/s41467-018-04760-1 OPEN Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role Shao-Ming Shen1, Yan Ji2, Cheng Zhang1, Shuang-Shu Dong2, Shuo Yang1, Zhong Xiong1, Meng-Kai Ge1, Yun Yu1, Li Xia1, Meng Guo1, Jin-Ke Cheng3, Jun-Ling Liu1,3, Jian-Xiu Yu1,3 & Guo-Qiang Chen1 Dysregulation of pre-mRNA alternative splicing (AS) is closely associated with cancers. However, the relationships between the AS and classic oncogenes/tumor suppressors are 1234567890():,; largely unknown. Here we show that the deletion of tumor suppressor PTEN alters pre-mRNA splicing in a phosphatase-independent manner, and identify 262 PTEN-regulated AS events in 293T cells by RNA sequencing, which are associated with significant worse outcome of cancer patients. Based on these findings, we report that nuclear PTEN interacts with the splicing machinery, spliceosome, to regulate its assembly and pre-mRNA splicing. We also identify a new exon 2b in GOLGA2 transcript and the exon exclusion contributes to PTEN knockdown-induced tumorigenesis by promoting dramatic Golgi extension and secretion, and PTEN depletion significantly sensitizes cancer cells to secretion inhibitors brefeldin A and golgicide A. Our results suggest that Golgi secretion inhibitors alone or in combination with PI3K/Akt kinase inhibitors may be therapeutically useful for PTEN-deficient cancers. 1 Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai 200025, China. 2 Institute of Health Sciences, Shanghai Institutes for Biological Sciences of Chinese Academy of Sciences and SJTU-SM, Shanghai 200025, China. -

Rattlesnake Genome Supplemental Materials 1 SUPPLEMENTAL
Rattlesnake Genome Supplemental Materials 1 1 SUPPLEMENTAL MATERIALS 2 Table of Contents 3 1. Supplementary Methods …… 2 4 2. Supplemental Tables ……….. 23 5 3. Supplemental Figures ………. 37 Rattlesnake Genome Supplemental Materials 2 6 1. SUPPLEMENTARY METHODS 7 Prairie Rattlesnake Genome Sequencing and Assembly 8 A male Prairie Rattlesnake (Crotalus viridis viridis) collected from a wild population in Colorado was 9 used to generate the genome sequence. This specimen was collected and humanely euthanized according 10 to University of Northern Colorado Institutional Animal Care and Use Committee protocols 0901C-SM- 11 MLChick-12 and 1302D-SM-S-16. Colorado Parks and Wildlife scientific collecting license 12HP974 12 issued to S.P. Mackessy authorized collection of the animal. Genomic DNA was extracted using a 13 standard Phenol-Chloroform-Isoamyl alcohol extraction from liver tissue that was snap frozen in liquid 14 nitrogen. Multiple short-read sequencing libraries were prepared and sequenced on various platforms, 15 including 50bp single-end and 150bp paired-end reads on an Illumina GAII, 100bp paired-end reads on an 16 Illumina HiSeq, and 300bp paired-end reads on an Illumina MiSeq. Long insert libraries were also 17 constructed by and sequenced on the PacBio platform. Finally, we constructed two sets of mate-pair 18 libraries using an Illumina Nextera Mate Pair kit, with insert sizes of 3-5 kb and 6-8 kb, respectively. 19 These were sequenced on two Illumina HiSeq lanes with 150bp paired-end sequencing reads. Short and 20 long read data were used to assemble the previous genome assembly version CroVir2.0 (NCBI accession 21 SAMN07738522). -

The Ultimated Edition to the Mountdomain
THEULTIMATED EDITIONUS 20170247762A1 TO THE MOUNTDOMAIN ( 19) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2017/ 0247762 A1 Heiman ( 43 ) Pub . Date: Aug . 31 , 2017 ( 54 ) COMPOSITIONS , METHODS AND USE OF Publication Classification SYNTHETIC LETHAL SCREENING (51 ) Int . CI. C120 1 /68 ( 2006 . 01 ) (71 ) Applicants : THE BOARD INSTITUTE INC ., C12N 15 / 113 ( 2006 .01 ) Cambridge, MA (US ) ; Massachusetts COZK 16 / 40 ( 2006 .01 ) Institute of Technology , Cambridge , A01K 67 /027 (2006 . 01 ) C12N 9 / 22 ( 2006 . 01 ) MA (US ) (52 ) U . S . CI. CPC . .. C12Q 1 /6883 (2013 . 01) ; A01K 67 /0278 (72 ) Inventor : Myriam Heiman , Newton , MA (US ) ( 2013 .01 ) ; C12N 9 / 22 ( 2013 .01 ) ; CO7K 16 / 40 ( 2013 .01 ) ; C12Y 111 /01009 ( 2013 . 01 ) ; C12N (21 ) Appl. No. : 15 /521 , 780 15 / 113 (2013 .01 ) ; AOIK 2267 / 0318 ( 2013 .01 ) ; C12Q 2600 / 118 ( 2013 . 01 ) ; C12Q 2600 / 158 (22 ) PCT Filed : Oct . 27 , 2015 (2013 .01 ) ; C120 2600 / 136 (2013 .01 ) ; C12N ( 86 ) PCT No. : PCT/ US2015 / 057567 2310 / 20 ( 2017 .05 ) ; CO7K 2317 / 34 (2013 .01 ) $ 371 ( C ) ( 1 ), (57 ) ABSTRACT Apr. 25 , 2017 The present invention generally relates to methods of iden ( 2 ) Date : tifying modulators of central nervous system diseases and the use of the modulators in treatment and diagnosis . The methods utilize a novel high throughput screen that includes Related U . S . Application Data injection of a library of barcoded viral vectors expressing (60 ) Provisional application No . 62/ 122, 686 , filed on Oct. shRNA ' s , CRISPR / Cas systems or cDNA ' s into animal 27 , 2014 . models of disease and detecting synthetic lethality . -

Loci Associated with Bone Strength in Laying Hens
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Epsilon Archive for Student Projects Faculty of Veterinary Medicine and Animal Science Department of Animal Breeding and Genetics Loci associated with bone strength in laying hens Biaty Raymond Examensarbete / Swedish University of Agricultural Sciences Master’s Thesis, 30 hp Department of Animal Breeding and Genetics 488 Uppsala 2016 Faculty of Veterinary Medicine and Animal Science Department of Animal Breeding and Genetics Loci associated with bone strength in laying hens Biaty Raymond Supervisors: D.J. De-Koning, SLU, Department of Animal Breeding and Genetics Anna Maria Johansson, SLU, Department of Animal Breeding and Genetics Examiner: Erling Strandberg, SLU, Department of Animal Breeding and Genetics Credits: 30 hp Course title: Degree project in Animal Science Course code: EX0556 Level: Advanced, A2E Place of publication: Uppsala Year of publication: 2016 Name of series: Examensarbete / Swedish University of Agricultural Sciences, Department of Animal Breeding and Genetics, 488 On-line publicering: http://epsilon.slu.se Key words: Osteoporosis, GWAS, Fracture, Genes, QTLs ABSTRACT One of the growing welfare concern in the layer industry is the high incidences of bone fracture. This is thought to result from reduction in bone strength due to osteoporosis which is exacerbated by environmental stresses and mineral deficiencies. Despite these factors however, the primary cause of bone weakness and the resulting fractures is believed to be genetic predisposition. In this study, we performed a genome-wide association study to identify with high reliability the loci associated with bone strength in laying hens. Genotype information and phenotype data were obtained from 752 laying hens belonging to the same pure line population. -

WO 2013/064702 A2 10 May 2013 (10.05.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2013/064702 A2 10 May 2013 (10.05.2013) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12Q 1/68 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, PCT/EP2012/071868 KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (22) International Filing Date: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 5 November 20 12 (05 .11.20 12) NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, (25) Filing Language: English TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (26) Publication Language: English ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 1118985.9 3 November 201 1 (03. 11.201 1) GB kind of regional protection available): ARIPO (BW, GH, 13/339,63 1 29 December 201 1 (29. 12.201 1) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, (71) Applicant: DIAGENIC ASA [NO/NO]; Grenseveien 92, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, N-0663 Oslo (NO). -

Tumors Normals
! ii! Dedications and acknowledgements ! The text of chapter three of this dissertation are a reprint of the material as it appears in Mazor T., Pankov A., et al., DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell, 2015. 28(3): p. 307-317 and Mazor T., Pankov A, Song J S, Costello J F. Intratumoral Heterogeneity of the Epigenome. Accepted at Cancer Cell, 2016. I would like to start by thanking the fantastic researchers that I had the great priviledge of working with throughout my graduate degree. I would specifically like to thank Tali Mazor, Julia Ye, and Jacob Freimer for sharing their wisdom and technical expertise to create an extraordinary collaborative environment that combined the experimental and computational aspects of genomics. The successful completion of our projects would not have been possible without their extensive knowledge. I would also like to expressed my gratitude to Dr. Robert Blelloch for his dedicated mentorship and ongoing support in helping to design the novel techniques used for our projects with his lab. A special thanks to Dr. Howard Rosen and Dr. John Kornak for providing continuing guidance on a non-genomics project to identify shrinking brain regions in dementia. They allowed me to push the boundaries of my learning and expand how I think about the applications of my work. I would especially like to thank the many researchers whose incredible skills have inspired me to become a better, more well-rounded scientist. I was incredibly priviledged to have Dr. Adam Olshen, Dr. -

Characterization of Five Transmembrane Proteins: with Focus on the Tweety, Sideroflexin, and YIP1 Domain Families
fcell-09-708754 July 16, 2021 Time: 14:3 # 1 ORIGINAL RESEARCH published: 19 July 2021 doi: 10.3389/fcell.2021.708754 Characterization of Five Transmembrane Proteins: With Focus on the Tweety, Sideroflexin, and YIP1 Domain Families Misty M. Attwood1* and Helgi B. Schiöth1,2 1 Functional Pharmacology, Department of Neuroscience, Uppsala University, Uppsala, Sweden, 2 Institute for Translational Medicine and Biotechnology, Sechenov First Moscow State Medical University, Moscow, Russia Transmembrane proteins are involved in many essential cell processes such as signal transduction, transport, and protein trafficking, and hence many are implicated in different disease pathways. Further, as the structure and function of proteins are correlated, investigating a group of proteins with the same tertiary structure, i.e., the same number of transmembrane regions, may give understanding about their functional roles and potential as therapeutic targets. This analysis investigates the previously unstudied group of proteins with five transmembrane-spanning regions (5TM). More Edited by: Angela Wandinger-Ness, than half of the 58 proteins identified with the 5TM architecture belong to 12 families University of New Mexico, with two or more members. Interestingly, more than half the proteins in the dataset United States function in localization activities through movement or tethering of cell components and Reviewed by: more than one-third are involved in transport activities, particularly in the mitochondria. Nobuhiro Nakamura, Kyoto Sangyo University, Japan Surprisingly, no receptor activity was identified within this dataset in large contrast with Diego Bonatto, other TM groups. The three major 5TM families, which comprise nearly 30% of the Departamento de Biologia Molecular e Biotecnologia da UFRGS, Brazil dataset, include the tweety family, the sideroflexin family and the Yip1 domain (YIPF) Martha Martinez Grimes, family. -

Intra-Genomic GC Heterogeneity in Sauropsids
Matsubara et al. BMC Genomics 2012, 13:604 http://www.biomedcentral.com/1471-2164/13/604 RESEARCH ARTICLE Open Access Intra-genomic GC heterogeneity in sauropsids: evolutionary insights from cDNA mapping and GC3 profiling in snake Kazumi Matsubara1,7*, Shigehiro Kuraku2,3, Hiroshi Tarui3,8, Osamu Nishimura3,4, Chizuko Nishida5, Kiyokazu Agata4, Yoshinori Kumazawa1 and Yoichi Matsuda6 Abstract Background: Extant sauropsids (reptiles and birds) are divided into two major lineages, the lineage of Testudines (turtles) and Archosauria (crocodilians and birds) and the lineage of Lepidosauria (tuatara, lizards, worm lizards and snakes). Karyotypes of these sauropsidan groups generally consist of macrochromosomes and microchromosomes. In chicken, microchromosomes exhibit a higher GC-content than macrochromosomes. To examine the pattern of intra-genomic GC heterogeneity in lepidosaurian genomes, we constructed a cytogenetic map of the Japanese four-striped rat snake (Elaphe quadrivirgata) with 183 cDNA clones by fluorescence in situ hybridization, and examined the correlation between the GC-content of exonic third codon positions (GC3) of the genes and the size of chromosomes on which the genes were localized. Results: Although GC3 distribution of snake genes was relatively homogeneous compared with those of the other amniotes, microchromosomal genes showed significantly higher GC3 than macrochromosomal genes as in chicken. Our snake cytogenetic map also identified several conserved segments between the snake macrochromosomes and the chicken microchromosomes. Cross-species comparisons revealed that GC3 of most snake orthologs in such macrochromosomal segments were GC-poor (GC3 < 50%) whereas those of chicken orthologs in microchromosomes were relatively GC-rich (GC3 ≥ 50%). Conclusion: Our results suggest that the chromosome size-dependent GC heterogeneity had already occurred before the lepidosaur-archosaur split, 275 million years ago. -

Diagnóstico Molecular Do Transtorno Do Espectro Autista Através Do Sequenciamento Completo De Exoma Molecular Diagnosis Of
Universidade de São Paulo Tatiana Ferreira de Almeida Diagnóstico molecular do transtorno do espectro autista através do sequenciamento completo de exoma Molecular diagnosis of autism spectrum disorder through whole exome sequencing São Paulo 2018 Universidade de São Paulo Tatiana Ferreira de Almeida Diagnóstico molecular do transtorno do espectro autista através do sequenciamento completo de exoma Molecular diagnosis of autism spectrum disorder through whole exome sequencing Tese apresentada ao Instituto de Biociências da Universidade de São Paulo, para a obtenção de Título de Doutor em Ciências, na Área de Biologia/Genética. Versão corrigida. Orientador(a): Maria Rita dos Santos Passos-Bueno São Paulo ii 2018 Dedico esta tese à ciência, e a todos os seus amantes. O, reason not the need! Our basest beggars Are in the poorest thing superfluous. Allow not nature more than nature needs, Man’s life is cheap as beast’s. Thou art a lady: If only to go warm were gorgeous, Why, nature needs not what thou gorgeous wear’st Which scarcely keeps thee warm. But, for true need- You heavens, give me that patience, patience I need! You see me here, you gods, a poor old man, As full of grief as age; wretched in both.” iii King Lear, William Shakespeare, 1608 Agradecimentos Agradeço em primeiro lugar ao meu grande amor Danilo, sem ele, há 11 anos, essa jornada não teria começado, a vida não teria me surpreendido e eu estaria de volta em casa, com todos os peixinhos do mar, e infeliz pelo resto da minha vida. Agradeço minha família, principalmente minha mãe Flávia e meu pai Airton por sacrificarem com mínimos protestos, as melhores horas que passaríamos juntos para que eu cumprisse meu trabalho. -
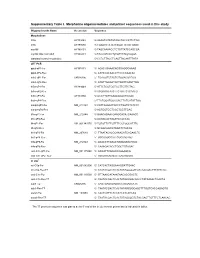
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN.