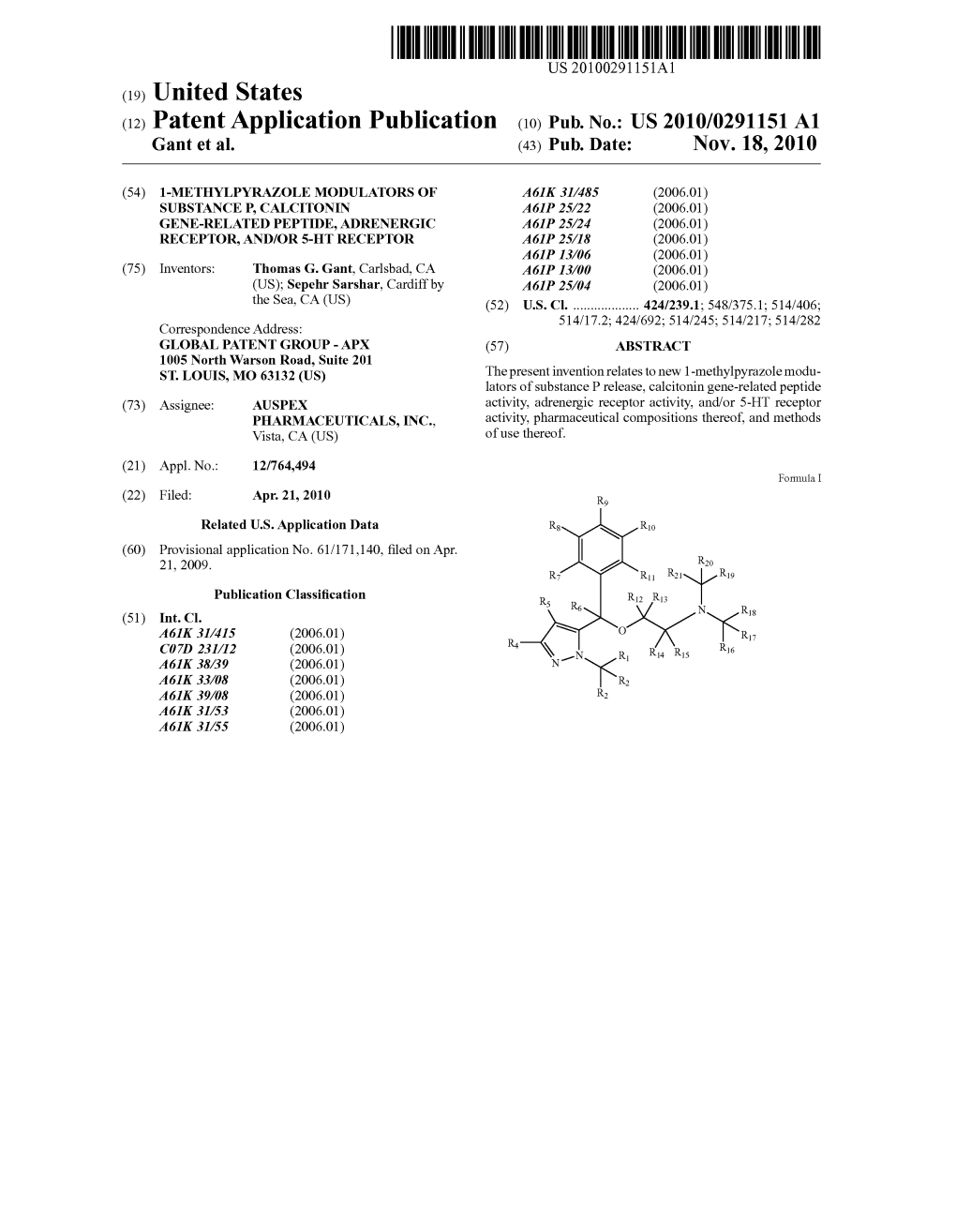
(12) Patent Application Publication (10) Pub. No.: US 2010/0291151 A1 Gant Et Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Discovering the Role of Autoinhibition in Kinetics Heterogeneity of Evoked Dopamine Responses
DISCOVERING THE ROLE OF AUTOINHIBITION IN KINETICS HETEROGENEITY OF EVOKED DOPAMINE RESPONSES TITLE PAGE by Han-Chen Wu B.S. Chemistry; B.A. Biochemistry, University of Washington, 2013 Submitted to the Graduate Faculty of the Kenneth P. Dietrich School of Arts and Sciences in partial fulfillment of the requirements for the degree of Master of Science University of Pittsburgh 2017 COMMITTEE UNIVERSITY OF PITTSBURGH DIETRICH SCHOOL OF ARTS AND SCIENCES This thesis was presented by Han-Chen Wu It was defended on November 15, 2016 and approved by Shigeru Amemiya, Associate Professor, Department of Chemistry Renã A.S. Robinson, Assistant Professor, Department of Chemistry Committee Chair: Adrian C. Michael, Professor, Department of Chemistry MEMBERSHIP PAGE ii Copyright © by Han-Chen Wu 2017 iii Adrian C. Michael DISCOVERING THE ROLE OF AUTOINHIBITION IN KINETICS HETEROGENEITY OF EVOKEDABSTRACT DOPAMINE PAGE RESPONSES Han-Chen Wu, M.S. University of Pittsburgh, 2017 Dopamine (DA) contributes to critical functions in the central nervous system. By introducing electrical stimulation in vivo at the rat’s medial forebrain bundle, evoked DA signals can be measured at a carbon fiber electrode with fast scan cyclic voltammetry (FSCV) in rat’s dorsal striatum. Previously, evidence suggested that the heterogeneity in evoked DA responses reflects the difference in inherent kinetics, which might play an important role physiologically. A restricted diffusion based numerical model produced great fit between simulations and observed responses for stimulation lengths no longer than 3 seconds, using 4 or sometimes 3 adjustable parameters. A study was designed to explore different supra-physiological stimuli and examine these evoked responses in the dorsal striatum after raclopride, a D2 antagonist drug, was given. -

Cizolirtine Citrate, an Effective Treatment for Symptomatic Patients with Urinary Incontinence Secondary T
EUROPEAN UROLOGY 56 (2009) 184–192 191 Editorial Comment on: Cizolirtine Citrate, an Effective the sample size was limited and placebo was chosen as the Treatment for Symptomatic Patients with Urinary comparator. The results achieved by cizolirtine seem to be Incontinence Secondary to Overactive Bladder: similar to antimuscarinic drugs currently used for OAB A Pilot Dose-Finding Study treatment. The different therapeutic mechanism suggests Florian May, Ricarda Bauer the use of cizolirtine both in patients nonresponsive to Department of Urology, Ludwig-Maximilians-University antimuscarinics and in combination with these drugs so as (LMU), Marchioninistr. 15, 81377 Munich, Germany to achieve a stronger effect. Subsequent studies including fl[email protected] a higher number of patients and a positive control treatment are necessary to get more information on this Antimuscarinic agents represent the first-line therapy for promising agent. overactive bladder (OAB). Objective clinical data and systematic reviews confer a high level of evidence and strong recommendations [1]. Many urologists, however, believe that pharmacologic management of OAB is not References satisfactory. There is no consensus on how long patients [1] Chapple C, Khullar V, Gabriel Z, Dooley JA. The effects of anti- should be treated, whether treatment should be contin- muscarinic treatment in overactive bladder: a systematic review uous, intermittent, or on demand, and why only relatively and meta-analysis. Eur Urol 2005;48:5–26. few patients remain on medication [2]. Pharmacologic [2] Hampel C. Long-term management of overactive bladder with research tries to answer these open questions and is looking antimuscarinic agents. Eur Urol Suppl 2007;6:432–7. -

DOPAC Dihydroxyphenylacetic Acid
NOTE TO USERS The original manuscript received by UMI contains pages with slanted print. Pages were microfilmed as received. This reproduction Is the best copy available UMI Université de Sherbrooke Time course and regional specificity of neurochemical changes in the rat brais following intra-accumbnl and intra-striatal injections of 6,7-ADTN (2-amino-6,7-dihydrory-l,2,3-tetrahydronaphthaIene) Jennifer M. Arnold Département de pharmacologie Mémoire présenté à la Faculté de Médecine en vue de l'obtention du grade de maître es sciences (M.Sc.) en Pharmacologie Juillet 1996 National Library Bibliothèque nationale du Canada Acquisitions and Acquisitions et Bibliographic Services services bibliographiques 395 Wellington Street 395. nie Wellington Ottawa ON KIA ON4 Ottawa ON KI A ON4 Canada Canada The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant a la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microfom, vendre des copies de cette thèse sous paper or electronic formats. la fome de microfiche/film, de reproduction sur papier ou sur format électronique. The author retauis ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts from it Ni la thèse ni des extraits substantiels may be printed or ohenirise de cellegcine doivent être imprimés reproduced without the -

(12) Patent Application Publication (10) Pub. No.: US 2003/0176365A1 Blass (43) Pub
US 200301.76365A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0176365A1 Blass (43) Pub. Date: Sep. 18, 2003 (54) NUTRITIONAL SUPPLEMENT FOR Publication Classification CEREBRAL METABOLIC INSUFFICIENCES (51) Int. Cl." ..................... A61K 31/715; A61K 31/525; A61K 31/70; A61K 31/51; (76) Inventor: John P. Blass, Bronxville, NY (US) A61K 31/19; A61K 31/355; A61K 31/198; A61K 31/353; A61K 31/375 Correspondence Address: (52) U.S. Cl. ............... 514/23: 514/53; 514/61; 514/557; Michael L. Goldman 514/574; 514/565; 514/456; NXON PEABODY LLP Clinton Square 514/458; 514/474; 514/251; P.O. BOX 31051 514/276; 514/355; 514/350 Rochester, NY 14603-1051 (US) (57) ABSTRACT (21) Appl. No.: 10/379,816 The present invention relates to a pharmaceutical composi (22) Filed: Mar. 4, 2003 tion which includes a Sugar and a Krebs cycle intermediate, or Salt thereof, or a precursor of a Krebs cycle intermediate. Krebs cycle intermediates include citric acid, aconitic acid, Related U.S. Application Data isocitric acid, C.-ketoglutaric, Succinic acid, fumaric acid, malic acid, and oxaloacetic acid, and mixtures thereof. (63) Continuation of application No. 09/529,091, filed on Precursors of Krebs cycle intermediates are compounds Oct. 20, 2000, now Pat. No. 6,537,969, filed as 371 of converted by the body to form a Krebs cycle intermediate. international application No. PCT/US98/18120, filed The present invention also relates to administration of the on Sep. 1, 1998. pharmaceutical composition to treat an individual for a disorder involving impaired mitochondrial function and to (60) Provisional application No. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0096296A1 Fikstad Et Al
US 2005.0096.296A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0096296A1 Fikstad et al. (43) Pub. Date: May 5, 2005 (54) PHARMACEUTICAL COMPOSITIONS WITH Related U.S. Application Data SYNCHRONIZED SOLUBLIZER RELEASE (63) Continuation of application No. 10/700,838, filed on (76) Inventors: David Fikstad, Salt Lake City, UT Nov. 3, 2003. (US); Srinivasan Venkateshwaran, Salt Lake City, UT (US); Publication Classification Chandrashekar Giliyar, Salt Lake City, UT (US); Feng-Jing Chen, Salt (51) Int. Cl." ..................... A61K 31/724; A61K 31/426; Lake City, UT (US); Mahesh V. Patel, A61K 31/401; A61K 31/366 Salt Lake City, UT (US) (52) U.S. Cl. ............................ 514/58; 514/369; 514/423; 514/460; 514/548; 514/458; Correspondence Address: 514/571 COOLEY GODWARD, LLP 3000 EL CAMINO REAL (57) ABSTRACT 5 PALO ALTO SQUARE PALO ALTO, CA 94.306 (US) Pharmaceutical compositions with Synchronized Solubilizer release as well as various methods associated there with, are (21) Appl. No.: 10/764,016 disclosed and described. More specifically, the aqueous Solubility of a drug is enhanced by Synchronized release of (22) Filed: Jan. 23, 2004 a solubilizer. Patent Application Publication May 5, 2005 Sheet 1 of 10 US 2005/0096296A1 C O C C N Vo V er (u/3m) AllqnOS SnoenbW OZeSOIO Patent Application Publication May 5, 2005 Sheet 2 of 10 US 2005/0096296A1 (%) eSeele JeZIqnOS %0·?–––i--|-----------|--------1–--------|--%0 84.9S#£Z|0 (?)QuiL (%) 9Seele IOZeSOILO Patent Application Publication May 5, 2005 Sheet -

(12) United States Patent (10) Patent No.: US 8,003,656 B2 Bakthavatchalam Et Al
US008.003656B2 (12) United States Patent (10) Patent No.: US 8,003,656 B2 Bakthavatchalam et al. (45) Date of Patent: Aug. 23, 2011 (54) 2-PHENOXY PYRIMIDINONE ANALOGUES WO WO-2005/0496O1 A1 6, 2005 WO WO-2005/049613 A1 6, 2005 WO WO-2005,12051.0 A1 12/2005 (75) Inventors: Rajagopal Bakshayatchalam, Madison, WO WO-2006/047516 A2 5, 2006 CT (US); Scott M. Capitosti, WO WO-2006, 120481 A2 11/2006 Middletown, CT (US); Jianjun Xu, WO WO2006,122200 A1 11, 2006 Branford, CT (US); Bertrand L. WO WO-2006, 122200 A1 11, 2006 s s WO WO-2007/0561.24 A2 5, 2007 Chenard, Waterford, CT (US); Manuka WO WO-2008,1566O7 12/2008 Ghosh, Madison, CT (US); Charles A. WO WO-2009/100403 8, 2009 Blum, Westbrook, CT (US) WO WO-2009/121.036 10/2009 (73) Assignee: Neurogen Corporation, Branford, CT OTHER PUBLICATIONS (US) Vippagunta, S.R., Adv. Drug. Delivery Rev. 2001, 48, p. 3-26.* The International Search Report and Written Opinion dated Sep. 8, (*) Notice: Subject to any disclaimer, the term of this 2008, from corresponding PCT Application No. PCT/US07/18654. patent is extended or adjusted under 35 Opposition filed in the name of Asociacion de la Industria U.S.C. 154(b) by 946 days. Farmaceutica Nacional Asifan dated Jul. 21, 2009, in corresponding Costa Rica Patent Application No. 10675 (English translation). (21) Appl. No.: 11/895,286 Johansen, M. E., et al., “TRPV1 Antagonists Elevate Cell Surface Populations of Receptor Protein and Exacerbate TRPV1-Mediated (22) Filed: Aug. -

Formulation for Controlled Release of Drugs by Combining Hydrophilic And
(19) & (11) EP 2 298 353 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.03.2011 Bulletin 2011/12 A61K 47/34 (2006.01) A61K 9/10 (2006.01) (21) Application number: 10012665.5 (22) Date of filing: 31.05.1996 (84) Designated Contracting States: • Kochinke, frank AT BE CH DE DK ES FI FR GB GR IE IT LI LU MC San Jose, CA 95148 (US) NL PT SE (74) Representative: Chung, Hsu Min (30) Priority: 02.06.1995 US 459134 Boult Wade Tennant Verulam Gardens (62) Document number(s) of the earlier application(s) in 70 Gray’s Inn Road accordance with Art. 76 EPC: London WC1X 8BT (GB) 04019466.4 / 1 477 187 96916903.6 / 0 831 912 Remarks: This application was filed on 01-10-2010 as a (71) Applicant: Allergan, Inc. divisional application to the application mentioned Irvine, CA 92612 (US) under INID code 62. (72) Inventors: • Wong, Vernon G. Menlo Park, CA 94025 (US) (54) Formulation for c ontrolled release of drugs by c ombining hydrophilic and hydr ophobic agents (57) An implant for sustained drug release compris- the implant; a release modulator at a concentration from ing: a pharmacologically acceptable biodegradable pol- 10 to 50 weight percent of the implant; wherein said ther- ymer which is degraded at the site of implantation, where- apeutically active agent is released within a therapeutic in said biodegradable polymer comprises at least about dosage which does not vary by more than about 100% 20 weight percent of the implant; a therapeutically active for a period of at least about 3 days. -

Vitro Evaluation of a Colon?Specific Controlled Release Drug Delivery
JPP 2008, 60: 1297–1303 ß 2008 The Authors Received April 14, 2008 Accepted June 23, 2008 Development and in-vitro evaluation of a colon-specific DOI 10.1211/jpp/60.10.0005 ISSN 0022-3573 controlled release drug delivery system Rahmat M. Talukder and Reza Fassihi Abstract The major challenges in targeting drug to various parts of the gastrointestinal tract include control of drug release with respect to its environment and transit time. These two variables should be taken into consideration in designing a rational colonic drug delivery system. To this end, a swelling matrix core containing pectin, hydroxypropyl methylcellulose (HPMC), microcrystalline cellulose and 5-aminosalicylic acid was developed. This was subjected to a dual coating operation: an inner pH-sensitive enteric and an outer semi-permeable membrane coat with a pore former. In-vitro dissolution studies were carried out in USP apparatus-I using sequential pH media. The first 2 h of dissolution studies were done in HCl buffer at pH 1.5, the next 2 h in pH 5.5 and, finally, in phosphate buffer at pH 6.8 with and without pectinolytic enzyme present. Less than 2% drug was released in the first 6 h and about 90% released in the following 12 h in a controlled manner. The stability studies of the coated systems were performed for 90 days under various conditions and it was found that drug release was not adversely affected. Results indicate that this delivery system has potential for site-specific delivery of drugs to the colon irrespective of transit time and rapid changes in the proximal pH of the gastrointestinal tract. -

Wo 2007/016496 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date (10) International Publication Number 8 February 2007 (08.02.2007) PCT WO 2007/016496 A2 (51) International Patent Classification: (74) Agents: KADLECEK, Ann et al.; Neurogen Corporation, A61K 31/551 (2006.01) A61K 31/4545 (2006.01) 35 Northeast Industrial Road, Branford, Connecticut 06405 A61K 31/55 (2006.01) A61K 31/454 (2006.01) (US). A61K 31/497 (2006.01) C07D 403/02 (2006.01) A61K 31/496 (2006.01) (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (21) International Application Number: AT, AU, AZ, BA, BB, BG, BR, BW, BY, BZ, CA, CH, CN, PCT/US2006/029761 CO, CR, CU, CZ, DE, DK, DM, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, HN, HR, HU, ID, IL, IN, IS, JP, (22) International Filing Date: 28 July 2006 (28.07.2006) KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, (25) Filing Language: English LU, LV,LY,MA, MD, MG, MK, MN, MW, MX, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, RO, RS, RU, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW (30) Priority Data: 60/704,722 2 August 2005 (02.08.2005) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): NEURO- GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, GEN CORPORATION [US/US]; Patent Department, 35 ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), Northeast Industrial Road, Branford, Connecticut 06405 European (AT,BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, (US). -

Neurochemical Investigation of Locally Induced Epilepsy And
Neurochemical Investigation of Locally Induced Epilepsy and Subsequent Oxidative Damage Using Microdialysis Sampling By Amanda Marie Furness B.S., B.A., Briar Cliff University Sioux City, IA, 51104 Submitted to the graduate degree program in Chemistry and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Chair: Susan M. Lunte Elias K. Michaelis Michael Johnson Bob Dunn Mario Rivera Date Defended: January, 31, 2017 The dissertation committee for Amanda Marie Furness certifies that this is the approved version of the following dissertation: Neurochemical Investigation of Locally Induced Epilepsy and Subsequent Oxidative Damage Using Microdialysis Sampling Chairperson: Susan M. Lunte Date Approved: February, 24, 2017 ii Abstract Amanda Marie Furness, Ph.D. R.N. Adams Institute for Bioanalytical Chemistry Department of Chemistry, January 2017 The University of Kansas The goal of this research was to develop and understand an anesthetized, multiple-seizure rat model for local epilepsy. Local seizures are not as well understood as global seizures due to their specificity and unpredictability. Furthermore, patients are diagnosed with epilepsy after experiencing two or more unprovoked seizures. In this model, two separate seizure episodes were induced by locally administering the epileptic agent 3-mercaptopropionic acid through a microdialysis probe to the CA1 region of the hippocampus. Upon development of the model, attenuation in glutamate release was observed in the second seizure stimulation. To investigate neurochemical and biochemical pathways which may be responsible for the glutamate diminution, the perfusion fluid was spiked with either glucose, lactate, or dihydrokainic acid. Additionally, as it is well known that high levels of extracellular glutamate can result in excitotoxicity, neuronal staining was performed to determine the neuronal viability after the induction of the first seizure. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0095995 A1 Wong Et Al
US 2003009.5995A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0095995 A1 Wong et al. (43) Pub. Date: May 22, 2003 (54) FORMULATION FOR CONTROLLED Related U.S. Application Data RELEASE OF DRUGS BY COMBINING HYDROPHILIC AND HYDROPHOBC (63) Continuation of application No. 09/160,635, filed on AGENTS Sep. 24, 1998, which is a continuation of application No. 08/459,134, filed on Jun. 2, 1995, now Pat. No. (76) Inventors: Vernon Wong, Rockville, MD (US); 5,869,079. Frank Kochinke, San Jose, CA (US) Publication Classification Correspondence Address: Mika Mayer (51) Int. Cl." ....................................................... A61K 9/00 Morrison & Foerster LLP (52) U.S. Cl. .............................................................. 424/426 755 Page Mill Road (57) ABSTRACT Palo Alto, CA 94.304-1018 (US) Combinations of hydrophilic and hydrophobic entities in a (21) Appl. No.: 10/327,018 biodegradable Sustained release implant are shown to modu late each other's rate of release. Formulations of a thera peutically active agent and modulator provide Substantially (22) Filed: Dec. 20, 2002 constant rate of release for an extended period of time. Patent Application Publication May 22, 2003 Sheet 1 of 4 US 2003/0095995 A1 Figure 1 | Patent Application Publication May 22, 2003. Sheet 2 of 4 US 2003/0095995 A1 Figure 2 a RT20-XTO32-40/40420 RT16-XTO29-50/50 . RRapp. 04 ugld per 100ug DDS B Time (d) Patent Application Publication May 22, 2003. Sheet 3 of 4 US 2003/0095995 A1 Figure 2 Time (hours) Patent Application Publication May 22, 2003. Sheet 4 of 4 US 2003/0095995 A1 st oo t CO () va lf S.N d an S. -

Madalina Victoria Natu Tavares.Phd Thesis.2011.Pdf
M˘ad˘alinaVictoria Natu Tavares Drug loading strategies and processing of polymer blends for intraocular controlled drug release application A Thesis submitted for the degree of Doctor in Chemical Engineering 2011 Abstract In 2010 there were 60.5 million people with glaucoma worldwide and this number is expected to increase to 79.6 million by 2020. In most glaucoma patients medical therapy consists of topical eyedrops. However, administration and compliance are often problematic. Surgical and laser treatment for glaucoma is traumatic for the patients, involving high costs and might require repetition of the procedure. Therefore the aim of the present work was the development of drug-eluting biodegradable implants designed to provide a localized and long-term (6 months to 2 years) sustained release of the drug that can be used in the treatment of glaucoma. The implant should degrade within the site of implantation, eliminating the need for further surgery. The implant should be introduced in the ophthalmologist office, under local anaesthesia, with high implications in patient recovery and costs. By delivering controlled amounts of drug the implant would be pharmacologically efficient and increase patient compliance. Several polymer processing and drug loading techniques were used in order to prepare controlled drug delivery systems (CDDS) for intraocular application. The chosen materials were poly("-caprolactone), PCL, because of its slow degradation, which makes it useful for long term delivery and poly(oxyethylene-b-oxypropylene- b-oxyethylene), Lu, because of its release modulation capacity. Moreover, they are both commercially available, inexpensive and well characterised polymers. Three drugs were incorporated: timolol maleate, acetazolamide and dorzolamide hydrochloride.