IL-33/ST2 Axis Promotes Glioblastoma Cell Invasion by Accumulating Tenascin-C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tenascin C: a Candidate for Chronic Inflammation?
RESEARCH HIGHLIGHTS RHEUMATOID ARTHRITIS Tenascin C: a candidate for chronic inflammation? After more than a decade of studying joint destruction after induction of during signal transduction) to block tenascin C, Kim Midwood and erosive arthritis—inflammatory cell FBG induction of IL-6. FBG also failed to colleagues have discovered a role for infiltration and synovial thickening induce cytokine synthesis in fibroblasts this extracellular matrix protein as an occurred, but tissue destruction and cell from Myd88–/– mice. Neutralizing endogenous ligand for Toll-like receptor death did not ensue. antibodies to TLR4 inhibited the (TLR) 4 that mediates persistent synovial Exogenous tenascin C induced tumor FBG-induced synthesis of TNF, IL-6 inflammation in arthritic joint disease. necrosis factor (TNF), interleukin and IL-8, and FBG could not induce these Tenascin C is normally only (IL)-6 and IL-8 in primary human cytokines in fibroblasts or macrophages expressed—transiently—in adult macrophages, and IL-6 in human from Tlr4–/– mice; furthermore FBG tissues in response to injury. However, fibroblasts. Tenascin C comprises several could not induce joint inflammation in in chronic inflammatory diseases, domains; the researchers established Tlr4–/–mice, indicating that FBG signals such as rheumatoid arthritis (RA), that only the ‘fibrinogen-like globe’ via TLR4. expression persists, particularly in the (FBG) domain was active in human The researchers plan to “…identify ways synovia, synovial fluid and cartilage. macrophages, synovial fibroblasts and to inhibit the proinflammatory action of This expression, together with the high an ex vivo model system of RA (synovial tenascin C in the hope that this may be homology of domains within tenascin C membranes from RA patients). -

Factory List to Demonstrate Our Pledge to Transparency
ASOS is committed to Fashion With Integrity and as such we have decided to publish our factory list to demonstrate our pledge to transparency. This factory list will be refreshed every three months to ensure that as we go through mapping it is continually up to date. This factory list does not include factories inherited from acquisitions made in February 2021. We are working hard to consolidate this supply base, and look forward to including these additional factories in our factory list once this is complete. Please see our public statement for our approach to the Topshop, Topman, Miss Selfridge and HIIT supply chains https://www.asosplc.com/~/media/Files/A/Asos-V2/reports-and- presentations/2021/asos-approach-to-the-topshop-topman-miss-selfridge-and-hiit- supply-chains.pdf Please direct any queries to [email protected] More information can be found in our ASOS Modern Slavery statement https://www.asosplc.com/~/media/Files/A/Asos- V2/ASOS%20Modern%20Slavery%20Statement%202020-21.pdf 31st May 2021 Number of Female Factory Name Address Line Country Department Male Workers Workers Workers 2010 Istanbul Tekstil San Ve Namik Kemal Mahallesi, Adile Nasit Bulvari 151, Sokak No. 161, B Turkey Apparel 150-300 53% 47% Dis Tic Ltd Sti Blok Kat1, Esenyurt, Istanbul, 34520 20th Workshop of Hong Floor 3, Building 16, Gold Bi Industrial, Yellow Tan Management Guang Yang Vacuum China Accessories 0-150 52% 48% District, Shenzhen, Guangdong, 518128 Technology Co., Ltd. (Nasihai) 359 Limited (Daisytex) 1 Ivan Rilski Street, Koynare, Pleven, 5986 -

Lee Luna.Pdf (3.311Mb)
RESPONSIBLE SILK – A PLAN OF ACTION FOR EILEEN FISHER A Project Paper Presented to the Faculty of the Graduate School of Cornell University in Partial Fulfillment of the Requirements for the Degree of Master of Professional Studies in Agriculture and Life Sciences Field of Global Development by Luna H. Lee August 2019 © 2019 Luna H. Lee ABSTRACT For apparel companies, agriculture is connected but often far removed. For EILEEN FISHER, being a socially conscious clothing company does not stop at the finished garment level. The company has been tracing its raw material supply chains for a few years, and is interested in expanding its human rights and environmental sustainability work to include the wellbeing of farmers and the land. In recent years, the company has started engaging suppliers and other external stakeholders at the agricultural level for cotton, wool, and man-made cellulosic. Silk is one of the top five fibers at EILEEN FISHER, representing 8% of its total materials in 2018, but one that is not well studied by the company. Grounded in literature review, this paper examines the different dimensions of the silk agricultural supply chain; the people, the land, and the silkworm. A review of the company’s silk supply chain revealed that 100% of its 2018 silk fiber comes from China, but little is known about its supply chain beyond the yarn spinner level. In collaboration with the company, a survey is conducted with its silk suppliers to trace the origin of silk cocoons within China. Findings indicates that the company’s silk originates from the provinces of Jiangsu and Guangxi. -
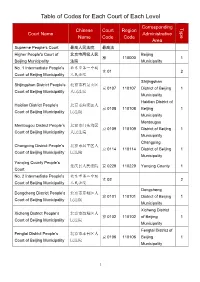
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Tenascin-C Induces Migration and Invasion Through JNK/C-Jun Signalling in Pancreatic Cancer
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 43), pp: 74406-74422 Research Paper Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer Jun Cai1, Shaoxia Du1, Hui Wang1, Beibei Xin1, Juan Wang1, Wenyuan Shen1, Wei Wei2, Zhongkui Guo1 and Xiaohong Shen1 1School of Medicine, Nankai University, Tianjin 300071, China 2Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin 300060, China Correspondence to: Xiaohong Shen, email: [email protected] Keywords: TNC, JNK/c-Jun, EMT, pancreatic cancer Received: December 28, 2016 Accepted: June 20, 2017 Published: August 10, 2017 Copyright: Cai et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Tenascin-C (TNC), a large extracellular matrix glycoprotein, has been reported to be associated with metastasis and poor prognosis in pancreatic cancer. However, the effects and mechanisms of TNC in pancreatic cancer metastasis largely remain unclear. We performed Transwell assays to investigate the effects of TNC on Capan-2, AsPC-1 and PANC-1 cells. In addition, western blot and RT-qPCR assays were used to examine potential TNC metastasis-associated targets, such as JNK/ c-Jun, Paxillin/FAK, E-cadherin, N-cadherin, Vimentin, and MMP9/2. Lastly, we utilized a variety of methods, such as immunofluorescence, gelatin zymography and immunoprecipitation, to determine the molecular mechanisms of TNC in pancreatic cancer cell motility. The present study showed that TNC induced migration and invasion in pancreatic cancer cells and regulated a number of metastasis-associated proteins, including the EMT markers, MMP9 and Paxillin. -

Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients with Stable Coronary Heart Disease
Supplementary Online Content Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. doi: 10.1001/jama.2016.5951 eTable 1. List of 1130 Proteins Measured by Somalogic’s Modified Aptamer-Based Proteomic Assay eTable 2. Coefficients for Weibull Recalibration Model Applied to 9-Protein Model eFigure 1. Median Protein Levels in Derivation and Validation Cohort eTable 3. Coefficients for the Recalibration Model Applied to Refit Framingham eFigure 2. Calibration Plots for the Refit Framingham Model eTable 4. List of 200 Proteins Associated With the Risk of MI, Stroke, Heart Failure, and Death eFigure 3. Hazard Ratios of Lasso Selected Proteins for Primary End Point of MI, Stroke, Heart Failure, and Death eFigure 4. 9-Protein Prognostic Model Hazard Ratios Adjusted for Framingham Variables eFigure 5. 9-Protein Risk Scores by Event Type This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Supplemental Material Table of Contents 1 Study Design and Data Processing ......................................................................................................... 3 2 Table of 1130 Proteins Measured .......................................................................................................... 4 3 Variable Selection and Statistical Modeling ........................................................................................ -

Spatial Distribution Pattern of Minshuku in the Urban Agglomeration of Yangtze River Delta
The Frontiers of Society, Science and Technology ISSN 2616-7433 Vol. 3, Issue 1: 23-35, DOI: 10.25236/FSST.2021.030106 Spatial Distribution Pattern of Minshuku in the Urban Agglomeration of Yangtze River Delta Yuxin Chen, Yuegang Chen Shanghai University, Shanghai 200444, China Abstract: The city cluster in Yangtze River Delta is the core area of China's modernization and economic development. The industry of Bed and Breakfast (B&B) in this area is relatively developed, and the distribution and spatial pattern of Minshuku will also get much attention. Earlier literature tried more to explore the influence of individual characteristics of Minshuku (such as the design style of Minshuku, etc.) on Minshuku. However, the development of Minshuku has a cluster effect, and the distribution of domestic B&Bs is very unbalanced. Analyzing the differences in the distribution of Minshuku and their causes can help the development of the backward areas and maintain the advantages of the developed areas in the industry of Minshuku. This article finds that the distribution of Minshuku is clustered in certain areas by presenting the overall spatial distribution of Minshuku and cultural attractions in Yangtze River Delta and the respective distribution of 27 cities. For example, Minshuku in the central and eastern parts of Yangtze River Delta are more concentrated, so are the scenic spots in these areas. There are also several concentrated Minshuku areas in other parts of Yangtze River Delta, but the number is significantly less than that of the central and eastern regions. Keywords: Minshuku, Yangtze River Delta, Spatial distribution, Concentrated distribution 1. -

Tenascin-C Expression Controls the Maturation of Articular Cartilage In
Gruber et al. BMC Res Notes (2020) 13:78 https://doi.org/10.1186/s13104-020-4906-8 BMC Research Notes RESEARCH NOTE Open Access Tenascin-C expression controls the maturation of articular cartilage in mice Bastian L. Gruber1, Michael J. Mienaltowski2,4, James N. MacLeod2, Johannes Schittny3, Stephanie Kasper1 and Martin Flück1,3* Abstract Objective: Expression of the de-adhesive extracellular matrix protein tenascin-C (TNC) is associated with the early postnatal development of articular cartilage which is both load-dependent and associated with chondrocyte diferen- tiation. We assessed morphological changes in the articular cartilage of TNC defcient mice at postnatal ages of 1, 4 and 8 weeks compared to age-matched wildtype mice. Results: Cartilage integrity was assessed based on hematoxylin and eosin stained-sections from the tibial bone using a modifed Mankin score. Chondrocyte density and cartilage thickness were assessed morphometrically. TNC expres- sion was localized based on immunostaining. At 8 weeks of age, the formed tangential/transitional zone of the articu- lar cartilage was 27% thicker and the density of chondrocytes in the articular cartilage was 55% lower in wildtype than the TNC-defcient mice. TNC protein expression was associated with chondrocytes. No relevant changes were found in mice at 1 and 4 weeks of age. The fndings indicate a role of tenascin-C in the post-natal maturation of the extracel- lular matrix in articular cartilage. This might be a compensatory mechanism to strengthen resilience against mechani- cal stress. Keywords: Tenascin C, Knock-out mouse, Articular cartilage, Cell density, Cartilage defect, Load, Adhesion Introduction adhesions [3–5]. -

Clinical Significance and Prognosis of Serum Tenascin-C in Patients with Sepsis
Yuan et al. BMC Anesthesiology (2018) 18:170 https://doi.org/10.1186/s12871-018-0634-1 RESEARCH ARTICLE Open Access Clinical significance and prognosis of serum tenascin-C in patients with sepsis Weifang Yuan1, Wei Zhang2, Xiaofang Yang1, Liyuan Zhou1, Ziwei Hanghua1 and Kailiang Xu1* Abstract Background: Tenascin-C is a pro-inflammatory glycoprotein with various biological functions. High expression of tenascin-C is found in inflammation, tissue remodeling, and autoimmune diseases. However, its expression and clinical significance in sepsis remain unclear. This study was designed to investigate the relationship between serum tenascin- C levels and disease severity and prognosis in patients with sepsis. Methods: A total of 167 patients with sepsis admitted to the ICU were enrolled. Lood samples were collected within 24 h of admission. Serum tenascin-C levels were measured by enzyme-linked immunosorbent assay (ELISA). Follow-up was performed to observe 30-day mortality. Results: Serum tenascin-C levels were significantly elevated in patients with sepsis compared with non-sepsis controls (P < 0.001). Serum tenascin-C levels were higher in nonsurvivors (58 cases) who died within 30 days (34.5%) compared to survivors (109 cases) (P < 0.001). In patients with sepsis, serum tenascin-C levels were significantly positively correlated with SOFA scores (P = 0.011), serum creatinine (P = 0.006), C-reactive protein (CRP) (P = 0.001), interleukin-6 (IL-6) (P < 0.001) , and tumor necrosis factor α (TNF-α)(P = 0.026). Logistic multivariate regression models showed that serum tenascin-C levels were independent contributor of 30-day mortality. Kaplan-Meier curves showed that septic patients with high levels of serum tenascin-C (≥56.9 pg/mL) had significantly higher 30-day mortality than those with lower serum tenascin-C (< 56.9 pg/mL) (P < 0.001). -

Tenascin-C: from Discovery to Structure-Function Relationships
OPINION published: 26 November 2020 doi: 10.3389/fimmu.2020.611789 Tenascin-C: From Discovery to Structure-Function Relationships Matthias Chiquet* Laboratory for Oral Molecular Biology, Department of Orthodontics and Dentofacial Orthopedics, University of Bern, Bern, Switzerland Keywords: recombinant protein, cDNA sequencing, electron microscopy, monoclonal antibodies, extracellular matrix, fibronectin, tenascin-C, rotary shadowing INTRODUCTION: HOW TO ISOLATE AN ECM PROTEIN IN 1980 Forty years ago, the tremendous complexity of extracellular matrix (ECM) was still largely an uncharted area, mainly because many of its components could only be solubilized by denaturing agents. Known were just five types of collagens, elastin, a couple of proteoglycans, and a few ECM glycoproteins, among them fibronectin, thrombospondin-1, and laminin-111 (1). The best studied was fibronectin (2), which became notorious for promoting specific cell adhesion to collagens. In parallel, the search for yet undetected large ECM glycoproteins continued. In 1981-82, Carter (3) observed several novel glycoproteins in human fibroblast ECM extracts. Among them, "GP250" was shown to be distinct from fibronectin but resisted isolation. However, between 1983-85 several Edited by: research groups independently discovered and characterized a similar ECM glycoprotein that later Kim Midwood, became known as tenascin-C (see below). Its subunits were comparable in size to fibronectin but University of Oxford, United Kingdom instead of dimers formed large (>106 kDa) disulfide-linked oligomers. In the following paragraphs, Reviewed by: the history and context of the individual discoveries of tenascin-C is briefly recounted. I then Richard P. Tucker, describe how a combination of methods available at the time lead to a detailed structural model of University of California, Davis, United States tenascin-C. -

Fibronectin and Tenascin-C: Accomplices in Vascular Morphogenesis During Development and Tumor Growth ELLEN VAN OBBERGHEN-SCHILLING*,1,2, RICHARD P
Int. J. Dev. Biol. 55: 511-525 doi: 10.1387/ijdb.103243eo www.intjdevbiol.com Fibronectin and tenascin-C: accomplices in vascular morphogenesis during development and tumor growth ELLEN VAN OBBERGHEN-SCHILLING*,1,2, RICHARD P. TUCKER3, FALK SAUPE4, ISABELLE GASSER4, BOTOND CSEH1,2, GERTRAUD OREND*,4,5,6 1CNRS UMR6543, IBDC, Nice, France, 2University of Nice-Sophia Antipolis, Nice, France, 3Cell Biology and Human Anatomy, University of California at Davis, USA, 4Inserm U682, Strasbourg, France, 5University of Strasbourg, UMR- S682, Strasbourg, France and 6Department of Molecular Biology, CHRU Strasbourg, Strasbourg, France ABSTRACT In addition to soluble factors, the extracellular matrix (ECM) also plays a vital role in normal vasculogenesis and in the pathological angiogenesis of many disease states. Here we will review what is known about the role of the ECM molecules fibronectin and tenascin-C in the vasculature and highlight a potential collaborative interplay between these molecules in develop- mental and tumorigenic angiogenesis. We will address the evolution of these modular proteins, their cellular interactions and how they become assembled into an insoluble matrix that impacts the assembly of other ECM proteins and the bioavailability of pro-angiogenic factors. The role of fibronectin and tenascin-C networks in tumor angiogenesis and metastasis will be described. We will elaborate on lessons learned about their role in vessel function from the functional ablation or the ectopic expression of both molecules. We will also elaborate on potential mechanisms of how fibronectin and tenascin-C affect cell adhesion and signaling that are relevant to angiogenesis. KEY WORDS: tumor angiogenesis, matrix assembly, fibronectin, tenascin-C Introduction blood vessels from preexisting vessels (Risau, 1997). -

The Evolution of Tenascins and Fibronectin
REVIEW Cell Adhesion & Migration 9:1-2, 22--33; January–April 2015; Published with License by Taylor & Francis Group, LLC The evolution of tenascins and fibronectin Josephine C Adams1, Ruth Chiquet-Ehrismann2,3, and Richard P Tucker4,* 1School of Biochemistry, University of Bristol; Bristol, UK; 2Friedrich Miescher Institute for Biomedical Research; Basel, Switzerland; 3University of Basel; Faculty of Science; Basel, Switzerland; 4Department of Cell Biology and Human Anatomy, University of California at Davis; Davis, CA USA Keywords: coelacanth, elephant shark, extracellular matrix, integrin, lamprey, phylogenomics Tenascins are extracellular matrix glycoproteins that act region that forms a coiled-coil, and most tenascins are believed to both as integrin ligands and as modifiers of fibronectin- exist as trimers or as hexamers formed from 2 trimers held integrin interactions to regulate cell adhesion, migration, together with disulfide bonds. Tenascins have several identified proliferation and differentiation. In tetrapods, both tenascins roles in cell adhesion and migration during development, tissue and fibronectin bind to integrins via RGD and LDV-type homeostasis and responses to disease or trauma, many of which fi tripeptide motifs found in exposed loops in their bronectin- are related to their ability to signal directly through integrin type III domains. We previously showed that tenascins receptors or by binding to the extracellular matrix glycoprotein appeared early in the chordate lineage and are represented fibronectin and influencing the way that fibronectin signals by single genes in extant cephalochordates and tunicates. through integrins (for review, see Chiquet-Ehrismann and Here we have examined the genomes of the coelacanth 1 Latimeria chalumnae, the elephant shark Callorhinchus milii as Tucker ).