Matthias Falk Aus Bonn
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Generation of Green Second Harmonic Radiation in LBO, Bibo, KTP, And
Generation of green second harmonic radiation in LBO, BiBO, KTP, and PPLN crystals using passively Q-switched sub-nanosecond microchip laser by Kostyantyn Sukhoy A Thesis submitted to the Faculty of Graduate Studies of The University of Manitoba in partial fulfillment of the requirements for the degree of Master of Science Department of Electrical and Computer Engineering University of Manitoba Winnipeg, Manitoba Canada Copyright © 2011 by Kostyantyn Sukhoy Author’s Declaration I hereby declare that I am the sole author of this thesis. This is not a finalized copy of my thesis. This is a nearly finalized draft of my thesis submitted to my examining committee for their consideration. I understand that an electronic copy of the finalized draft of my thesis may be made electronically available to the public. II Abstract A sub-nanosecond green laser source has big demand in such fields like spectroscopy, micromachining, fluorescence imaging, and laser displays. Most often green light is produced by frequency doubling of pulsed laser sources based on Nd3+-ion or Yb3+-ion doped gain media that oscillate in the near-IR range. For creating compact and relatively small source of green light suitable for broad type of applications a passively Q-switched Nd:YAG microchip laser (Teem Photonics) operating at 1064 nm with 6.9 kHz repetition rate was chosen. It delivers 560 ps long pulses with 10 µJ energy, corresponding to an average output power of 69 mW. Crystals of BiBO, KTP, LBO, and PPLN were chosen for frequency doubling. Main goal of this work was to study the characteristics of these crystals under similar experimental conditions and to select the most efficient one for this task. -

A Contribution to the Development of Wide Band-Gap Nonlinear Optical and Laser Materials
AN ABSTRACT OF THE THESIS OF Jennifer L. Stone-Sundberg for the degree of Doctor of Philosophy in Chemistry presented on December 3, 2001. Title: A Contribution to the Development of Wide Band-Gap Nonlinear Optical and Laser Materials Redacted for privacy Abstract approved: Douglas A. Keszler The primary focus of this work is on examining structure-property relationships of interest for high-power nonlinear optical and laser crystals. An intuitive and simply illustrated method for assessing the nonlinear optical potentialofstructurallycharacterizednoncentrosymmetricmaterialsis introduced.This method is applied to materials including common quartz and tourmaline and then extended to synthetic materials including borates, silicates, aluminates, and phosphates. Particularly,the contributions of symmetric tetrahedral and triangular anionic groups are inspected.It is shown that both types of groups significantly contribute to the optical frequency converting abilities of noncentrosymmetric crystals.In this study, several known materials are included as well as several new materials.The roles of the orientation, composition, and packing density of these anionic groups are also discussed. The structures and optical properties of the known materials BPO4, NaAlO2, LaCa4O(B03)3, and tourmaline; the new compounds Lao.8Y0.2Sc3(B03)4 and Ba2B10O17; and the laser host Sr3Y075Yb025(B03)3 are described. Copyright © December 3, 2001 by Jennifer Stone-Sundberg A Contribution to the Development of Wide Band-Gap Nonlinear Optical and Laser Materials by Jennifer L. Stone-Sundberg A THESIS Submitted to Oregon State University In partial fulfillment of The requirements for the Degree of Doctor of Philosophy Completed December 3, 2001 Commencement June 2002 Doctor of Philosophy dissertation of Jennifer L. -

Nonlinear Optical Frequency Conversion for Lasers in Space
Nonlinear Optical Frequency Conversion for Lasers in Space Diplomarbeit vorgelegt von Arne Potreck am Institut für Astronomie und Astrophysik, Eberhard-Karls Universität Tübingen und Institut für Technische Physik, Stuttgart Deutsches Zentrum für Luft- und Raumfahrt e.V. Juni 2014 F¨urAndrea Sophie Contents Deutsche Zusammenfassung 1 1 Introduction 3 1.1 Nonlinear frequency conversion onboard ADM-Aeolus . 4 1.2 Investigation of frequency conversion . 4 2 Background of the Diploma thesis 7 2.1 The ADM-Aeolus Mission . 7 2.1.1 Science . 9 2.1.2 The active instrument Aladin . 11 2.2 Nonlinear optical frequency conversion . 13 2.2.1 Introduction . 13 2.2.2 Theory of harmonic generation . 16 3 Choosing nonlinear crystals for the ADM-Aeolus Mission 23 3.1 Qualification of optics for space applications . 23 3.1.1 Bismuth Triborate . 25 3.1.2 Potassium Titanyl Phosphate . 26 3.1.3 Lithium Triborate . 27 4 Frequency conversion experiments 31 4.1 Experimental set-up . 31 4.1.1 Second harmonic generation . 32 4.1.2 Infrared-Lasersystem . 34 4.1.3 Mounting of the crystals . 35 4.1.4 Monitoring of the crystal's temperature . 36 4.1.5 Wavefront measurements . 37 4.1.6 Third harmonic generation . 43 4.1.7 Long term operation . 45 4.2 Results . 47 4.2.1 Qualification of the mount design . 47 4.2.2 Second harmonic generation . 50 v Contents 4.2.3 Third harmonic generation . 53 4.2.4 Longterm operation . 54 5 Laser calorimetric absorption measurements 59 5.1 Metrology of the absorption coefficient . -

Tilley R.J.D. Colour and the Optical Properties of Materials (Wiley, 2011
Colour and the Optical Properties of Materials An Exploration of the Relationship Between Light, the Optical Properties of Materials and Colour PROFESSOR RICHARD J. D. TILLEY Emeritus Professor, University of Cardiff, UK Colour and the Optical Properties of Materials Colour and the Optical Properties of Materials An Exploration of the Relationship Between Light, the Optical Properties of Materials and Colour PROFESSOR RICHARD J. D. TILLEY Emeritus Professor, University of Cardiff, UK This edition first published 2011 Ó 2011 John Wiley & Sons, Ltd Registered office John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, United Kingdom For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com. The right of the author to be identified as the author of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, withoutthe prior permission of the publisher. Wiley also publishes its books in a variety of electronic formats. Some content that appears in print may not be available in electronic books. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. -

Femtosecond Laser Pulses Amplification in Crystals
Review Femtosecond Laser Pulses Amplification in Crystals Razvan Dabu National Institute for Nuclear Physics and Engineering, Extreme Light Infrastructure-Nuclear Physics, Reactorului Street 30, 077125 Magurele, Romania; [email protected]; Tel.: +40-743147406 Received: 22 April 2019; Accepted: 1 July 2019; Published: 5 July 2019 Abstract: This paper describes techniques for high-energy laser pulse amplification in multi-PW femtosecond laser pulses. Femtosecond laser pulses can be generated and amplified in laser media with a broad emission spectral bandwidth, like Ti:sapphire crystals. By chirped pulse amplification (CPA) techniques, hundred-Joule amplified laser pulses can be obtained. Multi-PW peak-power femtosecond pulses are generated after recompression of amplified chirped laser pulses. The characteristics and problems of large bandwidth laser pulses amplification in Ti:sapphire crystals are discussed. An alternative technique, based on optical parametric chirped pulse amplification (OPCPA) in nonlinear crystals, is presented. Phase-matching conditions for broad bandwidth parametric amplification in nonlinear crystals are inferred. Ultra-broad phase matching bandwidth of more than 100 nm, able to support the amplification of sub-10 fs laser pulses, are demonstrated in nonlinear crystals, such as Beta Barium Borate (BBO), Potassium Dideuterium Phosphate (DKDP), and Lithium Triborate (LBO). The advantages and drawbacks of CPA amplification in laser crystals and OPCPA in nonlinear crystals are discussed. A hybrid amplification method, which combines low-medium energy OPCPA in nonlinear crystals with high energy CPA in large aperture laser crystals, is described. This technique is currently used for the development of 10-PW laser systems, with sub-20 fs pulse duration and more than 1012 intensity contrast of output femtosecond pulses. -
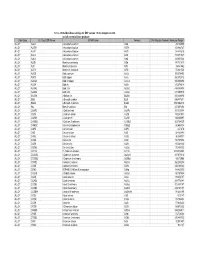
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

Nonlinear Crystals
NONLINEAR CRYSTALS Nonlinear Crystals LBO – LITHIUM TRIBORATE CRYSTALS NONLINEAR LBO is well suited for various nonlinear optical STANDARD SPECIFICATIONS appli cations: Flatness λ/8 at 633 nm frequency doubling and tripling of high ❯ Parallelism < 20 arcsec peak power pulsed Nd doped, Ti:Sapphire 10 – 5 scratch & dig Surface quality and Dye lasers (MIL-PRF-13830B) optical parametric oscillators (OPO) of both Perpendicularity < 5 arcmin ❯ Type 1 and Type 2 phase-match ing Angle tolerance < 30 arcmin LASER CRYSTALS LASER non-critical phase-matching for frequency Aperture tolerance ± 0.1 mm ❯ con ver sion of CW and quasi-CW radia tion. Clear aperture 90% of full aperture FEATURES 250 2.5 2.5 Type 1Type 2 Wide transparency region 200 ❯ 2 2 Broad Type 1 and Type 2 150 ❯ Type 1 Non-critical phase-matching (NCPM) range ❯ 100 1.5 1.5 XY plane YZ plane Small walk-off angle q=90 deg f=90 deg ❯ 50 High damage threshold ºC Temperature, ❯ 1 1 0 Type 2 TERAHERTZ CRYSTALS Wide acceptance angle Fundamental wavelength , µmλ ❯ High optical homogeneity -50 0.5 0.5 ❯ 0.8 1.0 1.2 1.4 1.6 1.8 90 60 30 0 0 30 60 90 Fundamental wavelength , µmλ Phase-matching angle, deg Phase-matching angle, deg NCPM SHG temperature dependance of LBO SHG tuning curves of LBO WE OFFER: STANDARD CRYSTALS LIST Crystals length ❯ Size, mm θ, deg φ, deg Coating Application Catalogue number Price, EUR up to 90 mm and 3x3x10 90 11.6 AR/AR @ 1064+532 nm SHG @ 1064 nm LBO-401 245 aper ture up to 3x3x15 90 11.6 AR/AR @ 1064+532 nm SHG @ 1064 nm LBO-402 325 CRYSTALS RAMAN 60 × 60 mm 4x4x10 -

∑ ∑ ∫ Short Range Order in Powder of Amorphous Anodic Yttrium Oxide
Crystallography Reports, Vol. 48, No. 4, 2003, pp. 531–535. Translated from Kristallografiya, Vol. 48, No. 4, 2003, pp. 583–587. Original Russian Text Copyright © 2003 by Aleshina, Loginova. CRYSTAL CHEMISTRY Short Range Order in Powder of Amorphous Anodic Yttrium Oxide L. A. Aleshina and S. V. Loginova Petrozavodsk State University, pr. Lenina 33, Petrozavodsk, 185640 Russia e-mail: [email protected] Received August 23, 2002 Abstract—The characteristics of the short-range order in a powder of amorphous yttrium oxide obtained by anodic oxidation are determined by the Finbak–Warren method. It is established that the first coordination num- ber equals seven. Amorphous oxide is built by irregular coordination polyhedra characteristic of the hexagonal and monoclinic modifications of yttrium oxide. © 2003 MAIK “Nauka/Interperiodica”. Yttrium oxide is widely used in planar optical is the convolution of the pair function Pij' (r) = devices for electroluminescent screens, in germanium- s max f (s)f (s)gÐ2(s)exp(Ð(α2 + σ 2 )s2)sin(sr )sin(sr)ds and silicon-based MIS varicaps and transistors, photo- ∫0 i j ij ij electric instruments, and nuclear technology. The oxide with the function of spreading of coordination spheres studied here is one of the most popular materials in in an amorphous material given in the form of a Gaus- micro- and optoelectronics and devices for displaying ()2 information. Thin films are also used as barriers, rr– ij Y2O3 sian G (r, r ) = Aexp–-------------------- . This representation efficient luminophor matrices, and dielectric interlay- ij ij σ 2 ers. ij allowed us to calculate the coordination numbers Nij by Amorphous Y2O3 oxide was studied in a limited the least squares method under the condition that number of publications [1]. -

Dfpub 63/198228
USPTO coversheet: Collaborative deferred-fee provisional patent application pilot program for COVID-19 invention, 85 Fed. Reg. 58038 (September 17, 2020) Identification number DFPUB_63198228 Date of filing 10/05/2020 Date available to public 10/28/2020 First inventor Ganio Title of invention Methods of treating infection and symptoms caused by SARS-CoV-2 using lithium Assignee (if any) -- Contact information Shawna Lemon 982 Trinity Road Raleigh, NC 27607 (919) 277-9100 [email protected] Attorney Docket No. 190709-00006(PR) Methods of Treating Infection and Symptoms Caused by SARS-CoV-2 Using Lithium Abstract The present inventive concept provides methods of using lithium to treat infection and symptoms caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS- CoV-2) and other pathogenic microbial organisms through direct application of lithium to areas of the respiratory tract. Lithium-based formulations and kits including the same for the treatment of respiratory infections and symptoms thereof are also provided. 12 Attorney Docket No. 190709-00006(PR) Methods of Treating Infection and Symptoms Caused by SARS-CoV-2 Using Lithium Field [0001] The present inventive concept provides methods of using lithium to treat infection and symptoms caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and other pathogenic microbial organisms affecting the respiratory tract. Background [0002] As disclosed in PCT/US2020/038736, the contents of which are incorporated herein by reference, formulations including lithium can be used for the prevention and treatment of inflammatory conditions, gout, joint disease and pain, and symptoms thereof. [0003] Mulligan et al., 1993 demonstrated that a murine antibody to human IL-8 had protective effects in inflammatory lung injury in rats. -

Trivalent Praseodymium-Doped Yttrium Lithium Fluoride Visible Lasers Pumped by Gallium Nitride Series Laser Diodes
Trivalent Praseodymium-doped Yttrium Lithium Fluoride Visible Lasers Pumped by Gallium Nitride Series Laser Diodes August 2018 Hiroki Tanaka Abstract This study deals with the demonstration of continuous-wave and passively Q-switched trivalent praseodymium (Pr3+)-doped solid-state lasers directly pumped by indium gallium nitride (InGaN)-based blue laser diodes 3+ (LDs). As the laser gain medium, Pr -doped yttrium lithium fluoride (LiYF4, YLF) was adopted in this study. Recent progress of InGaN-based blue LDs are remarkable, and nowadays, an output power of 5 W is available around 450 nm from a 9-mm CAN single emitter device. By utilizing the high power blue LDs, the pump power was increased up to 20 W, the highest pump level for visible Pr3+ lasers ever reported, and continuous output powers of 6.7 W and 3.7 W were obtained from Pr3+:YLF lasers operated at 640 and 607 nm, respectively. The onset of strong negative thermal lensing for π-polarization inhibited the scaling at 523 nm. The highest efficiency of 640-nm diode-pumped Pr3+:YLF laser was achieved by directly exciting the 479-nm line by a cyan-blue LD providing 1-W pump power. The highest slope efficiency of 62.9% was obtained. Using a fiber-coupled pump module provided >20 W pump power at 445 nm, the pump-limited scaling of an end-pumped Pr3+:YLF laser at 640 nm was demonstrated. The laser performance exhibited an onset of strong thermally-induced diffraction loss when high transmission output couplers were used. Using an output coupler of 1.1% transmission, an output power of 3.4 W was obtained with a slope efficiency of 25%. -

Crystals Selection Guide
CRYSTALS N ONLINEA Crystals R Selection Guide C R YSTALS L ASE R C R YSTALS See page NONLINEAR CRYSTALS 2.3–2.17 Lithium Triborate – LBO 2.3 Beta Barium Borate – BBO 2.5 Potassium Dideuterium Phosphate – KDP, DKDP 2.7 T E R Potassium Titanyl Phosphate – KTP 2.9 AHE Potassium Titanyle Arsenate – KTA 2.11 R TZ Lithium Iodate – LiIO3 2.12 CR YSTALS Infrared Nonlinear Crystals 2.13 Ultrathin Nonlinear Crystals 2.16 RAMAN C See page LASER CRYSTALS 2.18–2.21 R Neodymium Doped Yttrium Aluminium Garnet – Nd:YAG 2.18 YSTALS Yb-Doped Potassium Gadolinium Tungstate – Yb:KGW, Yb:KYW 2.19 Nd-Doped Potassium Gadolinium Tungstate – Nd:KGW 2.20 Titanium Doped Sapphire – Ti:Sapphire 2.20 Lead Thiogallate with Dysprosium Ions Co-doped 3+ 2.21 by Alkali Metals – Dy :PbGa2S4 POSITIONE RS & H OLDE RS See page CR TERAHERTZ CRYSTALS 2.22–2.24 YSTAL YSTAL Organic Terahertz Crystals – DSTMS, DAST, OH1 2.22 O Semiconductor Terahertz Crystals – GaSe, ZnTe 2.24 VENS 2.1 EKSMA OPTICS • Tel.: +370 5 272 99 00 • Fax: +370 5 272 92 99 • [email protected] • www.eksmaoptics.com CRYSTALS SELECTion GuidE YSTALS R C R ONLINEA N See page RAMAN CRYSTALS 2.25–2.26 Crystals for Stimulated Raman Scattering – KGW, Ba(NO3)2 2.25 2+ 4+ Passive Q-switching Crystals – Co :MgAl2O4, Cr :YAG 2.26 YSTALS R C R ASE L YSTALS CR TZ R See page AHE R E POSITIONERS & HOLDERS 2.27–2.29 T Ring Holders for Nonlinear Crystals 830-0001 2.27 Positioning Mount 840-0056-11 2.28 Kinematic Positioning Mount 840-0193 2.28 Positioning Mount for Nonlinear Crystal Housing 840-0199 2.29 YSTALS R RAMAN -

NLO Crystals CASTECH
R NLO Crystals CASTECH Lithium Triborate (LiB3O5, LBO) Introduction Lithium Triborate (LiB3O5 or LBO) is an excellent nonlinear optical crystal discovered and developed by FIRSM, CAS (Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences). Fujian CASTECH Crystals, Inc. (CASTECH) has the exclusive rights to produce, manufacture and market the patented LBO crystal and its NLO devices. The patent number is 4,826,283 in USA, 2023845 in Japan and 88 1 02084.2 in China. CASTECH’s LBO is featured by • broad transparency range from 160nm to 2600nm (see Figure 1); • high optical homogeneity (δn≈10-6/cm) and being free of inclusion; • relatively large effective SHG coefficient (about three times that of KDP); • high damage threshold; • wide acceptance angle and small walk-off; • type I and type II non-critical phase matching (NCPM) in a wide wavelength range; • spectral NCPM near 1300nm. CASTECH offers • strict quality control; • large crystal size up to 30x30x30mm3 and maximum length of 60mm; • AR-coating, mounts and re-polishing services; • a large quantity of crystals in stock; • fast delivery(10 days for polished only, 15 days for AR-coated). Table 1. Chemical and Structural Properties Crystal Structure Orthorhombic, Space group Pna21, Point group mm2 Lattice Parameter a=8.4473Å, b=7.3788Å, c=5.1395Å, Z=2 Melting Point About 834°C Mohs Hardness 6 Density 2.47 g/cm3 Thermal Conductivity 3.5W/m/K -5 -5 -5 Thermal Expansion Coefficients αx=10.8x10 /K, αy= -8.8x10 /K, αz=3.4x10 /K 1 Tel:+86 591 83710533 E-mail:[email protected] http://www.castech.com R CASTECH NLO Crystals Table 2.