Comprehensive Proteomic Analysis of the Human Spliceosome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subcellular RNA Profiling Links Splicing and Nuclear DICER1 to Alternative Cleavage and Polyadenylation
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation Jonathan Neve1#, Kaspar Burger2#, Wencheng Li3, Mainul Hoque3, Radhika Patel1, Bin Tian3, Monika Gullerova2* and Andre Furger1* Affiliations: 1 Department of Biochemistry, South Parks Road, University of Oxford, OX1 3QU, UK 2 Sir William Dunn School of Pathology, South Parks Road, University of Oxford, OX1 3RE, UK 3Department of Biochemistry and Molecular Biology, Rutgers New Jersey Medical School, Newark, NJ 07103, USA *Corresponding authors: Andre Furger Email: [email protected] Monika Gullerova Email: [email protected] # authors contributed equally Running Title: APA and subcellular fractionation Key words: alternative polyadenylation, mRNA, subcellular fractionation, DICER1 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Alternative cleavage and polyadenylation (APA) plays a crucial role in the regulation of gene expression across eukaryotes. Although APA is extensively studied, its regulation within cellular compartments and its physiological impact remains largely enigmatic. Here, we employed a rigorous subcellular fractionation approach to compare APA profiles of cytoplasmic and nuclear RNA fractions from human cell lines. This approach allowed us to extract APA isoforms that are subjected to differential regulation and provided us with a platform to interrogate the molecular regulatory pathways that shape APA profiles in different subcellular locations. Here we show that APA isoforms with shorter 3’UTRs tend to be overrepresented in the cytoplasm and appear to be cell type specific events. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

Molecular Basis for the Distinct Cellular Functions of the Lsm1-7 and Lsm2-8 Complexes
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.22.055376; this version posted April 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Molecular basis for the distinct cellular functions of the Lsm1-7 and Lsm2-8 complexes Eric J. Montemayor1,2, Johanna M. Virta1, Samuel M. Hayes1, Yuichiro Nomura1, David A. Brow2, Samuel E. Butcher1 1Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, USA. 2Department of Biomolecular Chemistry, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA. Correspondence should be addressed to E.J.M. ([email protected]) and S.E.B. ([email protected]). Abstract Eukaryotes possess eight highly conserved Lsm (like Sm) proteins that assemble into circular, heteroheptameric complexes, bind RNA, and direct a diverse range of biological processes. Among the many essential functions of Lsm proteins, the cytoplasmic Lsm1-7 complex initiates mRNA decay, while the nuclear Lsm2-8 complex acts as a chaperone for U6 spliceosomal RNA. It has been unclear how these complexes perform their distinct functions while differing by only one out of seven subunits. Here, we elucidate the molecular basis for Lsm-RNA recognition and present four high-resolution structures of Lsm complexes bound to RNAs. The structures of Lsm2-8 bound to RNA identify the unique 2′,3′ cyclic phosphate end of U6 as a prime determinant of specificity. In contrast, the Lsm1-7 complex strongly discriminates against cyclic phosphates and tightly binds to oligouridylate tracts with terminal purines. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Mrna Turnover Philip Mitchell* and David Tollervey†
320 mRNA turnover Philip Mitchell* and David Tollervey† Nuclear RNA-binding proteins can record pre-mRNA are cotransported to the cytoplasm with the mRNP. These processing events in the structure of messenger proteins may preserve a record of the nuclear history of the ribonucleoprotein particles (mRNPs). During initial rounds of pre-mRNA in the cytoplasmic mRNP structure. This infor- translation, the mature mRNP structure is established and is mation can strongly influence the cytoplasmic fate of the monitored by mRNA surveillance systems. Competition for the mRNA and is used by mRNA surveillance systems that act cap structure links translation and subsequent mRNA as a checkpoint of mRNP integrity, particularly in the identi- degradation, which may also involve multiple deadenylases. fication of premature translation termination codons (PTCs). Addresses Cotransport of nuclear mRNA-binding proteins with mRNA Wellcome Trust Centre for Cell Biology, ICMB, University of Edinburgh, from the nucleus to the cytoplasm (nucleocytoplasmic shut- Kings’ Buildings, Edinburgh EH9 3JR, UK tling) was first observed for the heterogeneous nuclear *e-mail: [email protected] ribonucleoprotein (hnRNP) proteins. Some hnRNP proteins †e-mail: [email protected] are stripped from the mRNA at export [1], but hnRNP A1, Current Opinion in Cell Biology 2001, 13:320–325 A2, E, I and K are all exported (see [2]). Although roles for 0955-0674/01/$ — see front matter these hnRNP proteins in transport and translation have been © 2001 Elsevier Science Ltd. All rights reserved. reported [3•,4•], their affects on mRNA stability have been little studied. More is known about hnRNP D/AUF1 and Abbreviations AREs AU-rich sequence elements another nuclear RNA-binding protein, HuR, which act CBC cap-binding complex antagonistically to modulate the stability of a range of DAN deadenylating nuclease mRNAs containing AU-rich sequence elements (AREs) DSEs downstream sequence elements (reviewed in [2]). -

RNA Components of the Spliceosome Regulate Tissue- and Cancer-Specific Alternative Splicing
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing Heidi Dvinge,1,2,4 Jamie Guenthoer,3 Peggy L. Porter,3 and Robert K. Bradley1,2 1Computational Biology Program, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 2Basic Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 3Human Biology Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA Alternative splicing of pre-mRNAs plays a pivotal role during the establishment and maintenance of human cell types. Characterizing the trans-acting regulatory proteins that control alternative splicing has therefore been the focus of much research. Recent work has established that even core protein components of the spliceosome, which are required for splicing to proceed, can nonetheless contribute to splicing regulation by modulating splice site choice. We here show that the RNA components of the spliceosome likewise influence alternative splicing decisions. Although these small nuclear RNAs (snRNAs), termed U1, U2, U4, U5, and U6 snRNA, are present in equal stoichiometry within the spliceosome, we found that their relative levels vary by an order of magnitude during development, across tissues, and across cancer samples. Physiologically relevant perturbation of individual snRNAs drove widespread gene-specific differences in alternative splic- ing but not transcriptome-wide splicing failure. Genes that were particularly sensitive to variations in snRNA abundance in a breast cancer cell line model were likewise preferentially misspliced within a clinically diverse cohort of invasive breast ductal carcinomas. -

The Reciprocal Regulation Between Splicing and 3-End Processing
Advanced Review The reciprocal regulation between splicing and 30-end processing Daisuke Kaida* Most eukaryotic precursor mRNAs are subjected to RNA processing events, including 50-end capping, splicing and 30-end processing. These processing events were historically studied independently; however, since the early 1990s tremendous efforts by many research groups have revealed that these processing factors interact with each other to control each other’s functions. U1 snRNP and its components negatively regulate polyadenylation of precursor mRNAs. Impor- tantly, this function is necessary for protecting the integrity of the transcriptome and for regulating gene length and the direction of transcription. In addition, physical and functional interactions occur between splicing factors and 30-end processing factors across the last exon. These interactions activate or inhibit spli- cing and 30-end processing depending on the context. Therefore, splicing and 30- end processing are reciprocally regulated in many ways through the complex protein–protein interaction network. Although interesting questions remain, future studies will illuminate the molecular mechanisms underlying the recipro- cal regulation. © 2016 The Authors. WIREs RNA published by Wiley Periodicals, Inc. How to cite this article: WIREs RNA 2016, 7:499–511. doi: 10.1002/wrna.1348 INTRODUCTION regulate each other on the transcription apparatus. In addition, because these events are carried out co- n eukaryotic cells, most precursor mRNAs (pre- transcriptionally in most cases, such factors can exist ImRNAs) consist of protein-coding regions, exons, on pre-mRNA just after synthesis of the binding sites 1,2 and intervening regions, introns. The pre-mRNAs of processing factors, suggesting that the factors are subjected to splicing to remove introns and to affect each other on the pre-mRNA. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins
Biomolecules 2015, 5, 1441-1466; doi:10.3390/biom5031441 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins Rebecca Bish 1,†, Nerea Cuevas-Polo 1,†, Zhe Cheng 1, Dolores Hambardzumyan 2, Mathias Munschauer 3, Markus Landthaler 3 and Christine Vogel 1,* 1 Center for Genomics and Systems Biology, Department of Biology, New York University, 12 Waverly Place, New York, NY 10003, USA; E-Mails: [email protected] (R.B.); [email protected] (N.C.-P.); [email protected] (Z.C.) 2 The Cleveland Clinic, Department of Neurosciences, Lerner Research Institute, 9500 Euclid Avenue, Cleveland, OH 44195, USA; E-Mail: [email protected] 3 RNA Biology and Post-Transcriptional Regulation, Max-Delbrück-Center for Molecular Medicine, Berlin-Buch, Robert-Rössle-Str. 10, Berlin 13092, Germany; E-Mails: [email protected] (M.M.); [email protected] (M.L.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-212-998-3976; Fax: +1-212-995-4015. Academic Editor: André P. Gerber Received: 10 May 2015 / Accepted: 15 June 2015 / Published: 15 July 2015 Abstract: DDX6 (p54/RCK) is a human RNA helicase with central roles in mRNA decay and translation repression. To help our understanding of how DDX6 performs these multiple functions, we conducted the first unbiased, large-scale study to map the DDX6-centric protein-protein interactome using immunoprecipitation and mass spectrometry. Using DDX6 as bait, we identify a high-confidence and high-quality set of protein interaction partners which are enriched for functions in RNA metabolism and ribosomal proteins. -

PLRG1 Antibody Cat
PLRG1 Antibody Cat. No.: 46-203 PLRG1 Antibody 46-203 (5ug/ml) staining of paraffin embedded Human Prostate. Steamed antigen retrieval with citrate buffer pH 6, AP-staining. Specifications HOST SPECIES: Goat SPECIES REACTIVITY: Human, Mouse HOMOLOGY: Expected Species Reactivity based on sequence homology: Rat, Dog, Pig, Cow IMMUNOGEN: The immunogen for this antibody is: C-PVSWKPEIIKRKRF TESTED APPLICATIONS: ELISA, IHC, WB Peptide ELISA: antibody detection limit dilution 1:16000.Western Blot:Approx 60kDa band observed in nuclear lysates of cell lines HEK293 and NIH3T3 (calculated MW of 57.2kDa according to Human NP_002660.1 and 56.9kDa according to Mouse APPLICATIONS: NP_058064.2). Recommended concentration: 0.1-0.3ug/ml. Primary incubation was 1 hour.Immunohistochemistry:Paraffin embedded Human Prostate. Recommended concentration: 5ug/ml. September 28, 2021 1 https://www.prosci-inc.com/plrg1-antibody-46-203.html This antibody is expected to recognise isoform 1 (NP_002660.1) and isoform 2 SPECIFICITY: (NP_001188493.1). POSITIVE CONTROL: 1) Cat. No. 1405 - Mouse Kidney Tissue Lysate Properties Purified from goat serum by ammonium sulphate precipitation followed by antigen PURIFICATION: affinity chromatography using the immunizing peptide. CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid Supplied at 0.5 mg/ml in Tris saline, 0.02% sodium azide, pH7.3 with 0.5% bovine serum BUFFER: albumin. Aliquot and store at -20°C. Minimize freezing and thawing. CONCENTRATION: 500 ug/mL STORAGE CONDITIONS: Aliquot and store at -20˚C. -
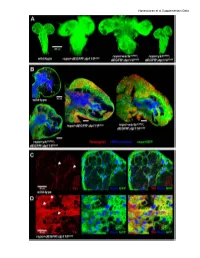
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells.