Air' Igiararle-... '
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carbon Dioxide and Chemical Looping: Current Research Trends Lukas C
Review Cite This: Ind. Eng. Chem. Res. XXXX, XXX, XXX−XXX pubs.acs.org/IECR 110th Anniversary: Carbon Dioxide and Chemical Looping: Current Research Trends Lukas C. Buelens, Hilde Poelman, Guy B. Marin, and Vladimir V. Galvita* Laboratory for Chemical Technology, Ghent University, Technologiepark 125, B-9052 Ghent, Belgium *S Supporting Information ABSTRACT: Driven by the need to develop technologies for converting CO2, an extraordinary array of chemical looping based process concepts has been proposed and researched over the past 15 years. This review aims at providing first a historical context of the molecule CO2, which sits at the center of these developments. Then, different types of chemical looping related to CO2 are addressed, with attention to process concepts, looping materials, and reactor configu- rations. Herein, focus lies on the direct conversion of carbon dioxide into carbon monoxide, a process deemed to have economic potential. 1. THE DISCOVERY OF CARBON DIOXIDE designate CO2 and CO probably occurred only after the Carbon dioxide was probably discovered around 1640 by Jan foundation of the International Union on Pure and Applied Baptist van Helmont, who named it spiritus sylvestre.1,2 When Chemistry (IUPAC) in 1919. burning a piece of wood, van Helmont noticed that the mass of 1,2 2. CARBON DIOXIDE AND THE EARTH’S CLIMATE the ash was considerably less than that of the original wood. He argued that a gas had volatilized from the wood.1,2 Over a In the 1820s, Joseph Fourier made heat transfer calculations hundred years later, around 1750, Joseph Black looked into based on knowledge of that time−between the Sun and the 9,10 carbon dioxide released by magnesium carbonate upon heating Earth. -

Materials and Chemical Sciences Division Lawrence Berkeley Laboratory • University of California ONE CYCLOTRON ROAD, BERKELEY, CA 94720 • (415) 486-4755
RECEI VED LBL-27307 c: ~ LAWRE~lCE n" f'' ;:.:v L.l\808!l.TORY 13 E,,.... L~· FEB 8 '1990 Center for Advanced Materials L Bi·\/~fN AND CAM ------DOCUMENTS SECTION To be published as a chapter in Alkali Adsorption on J"' Metals and Semiconductors, H.P. Bonzel, A.M. Bradshaw, and G. Ertl, Eds., Elsevier Science Publishers, BV, Amsterdam, The Netherlands, 1989 Alkali Metals as Structure and Bonding Modifiers of Transition l'VIetal Catalysts G.A. Somorjai and E.L. Garfunkel June 1989 TWO-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. ,. / ' . Materials and Chemical Sciences Division Lawrence Berkeley Laboratory • University of California ONE CYCLOTRON ROAD, BERKELEY, CA 94720 • (415) 486-4755 Prepared for the U.S. Department of Energy under Contract DE-AC03-76SF00098 DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. -

4923: 70-614 DATE 6 February 1970 W
CR- 72 963 10-047-009 (REV. 6/63) DIVISION Power Systems TM 4923: 70-614 DATE 6 February 1970 w. 0. 1139-78-2000 TECHNl CAL MEMO RAN DUM AUTHOR(S): F. H. Cassidy Oxides in NaK ABSTRACT The eutectic alloy of sodium and potassium (NaK) is very oxygen-reactive. A review was made of how the NaK and the SNAP-8 system are affected; the sources of oxygen contamination in a NaK system; the solubility of the oxide; the effects of the oxide in respect to the system and on the NaK; and oxide control methods. KEY WORDS: cold trapping, crystallization, hot trapping, mass transfer, NaK, oxides, potassium superoxide, sodium monoxide, solubility APPROVED: DEPARTMENT HEAD __ H. Derow NOTE: The information in this document is subject to revision as analysis progresses and additional data are acquired. AEROJ ET-GEN ERAL CORPORATION .. TABLE OF CONTENTS hge I. summary 1 11. Introduction 1 111. Alkali &tal Oxides 1 A. Sources of Oxygen 3 B. Solubility and Crystallization 4 C. The Effect of Oxygen on a Liquid Alkali Metal System 5 IV. Control of Alkali Metal Oxides 7 A. Operational Procedures to Prevent Contamination 7 B. Purification Techniques During Loop Opration 8 C. Other Inline Monitoring Devices 9 References 10 Figures Periodic Chart of the Elements 1 Binary Phase Diagram: Sodium/Potassium 2 Free Energy of Formation vs . Temperature for Selected Oxide 3 Equilibrium Curve of Temperature vs . Concentration of Sodium Monoxide, 4 Na20, as Oxygen Schematic of Purification System Pluggings Loop and Plugging Valve 5 > sc I ,* +- . Tab le Oxide of SodZum and Potassium 1 Changes of Physical Properties of Eutectic NaK Caused by Sodium 2 Depletion Appendix Explosives Incident No. -

Platinum Metals Review
UK ISSN 0032-1400 PLATINUM METALS REVIEW A quarterly survey of research on the platinum metals and of developments in their application in industry VOL. 35 JANUARY 1991 NO. 1 Contents Platinum in High Temperature Superconductor Technology 2 Polymetallic Activation 10 Advances in the Study of Platinum Group Elements 16 A Means to a Cleaner Environment 17 Quasicrystals in Rapidly Solidified Alloys 21 Grove Fuel Cell Symposium 21 The Photogeneration of Hydrogen 22 Cleavage in Iridium Crystals 23 Metal-Hydrogen Systems 24 Palladium Membrane Reactors 27 Isomers of Platinum(I1) and Palladium(1I) Complexes 28 Magneto-Optical Recording Materials 31 Osmium and Ruthenium Complexes 31 Abstracts 32 New Patents 47 Communications should be addressed to The Editor, Platinum Metals Review Johnson Matthey Public Limited Company, Hatton Garden, London EClN 8EE Platinum in High Temperature Superconductor Technology LITERATURE SURVEY SUGGESTS POTENTIAL USES By E. F. Maher Johnson Matthey Technology Centre High temperature superconducting oxides are being subjected to intensive investigation, designed to establish the basic mechanisms governing their superconductivity and to enable their electrical and mechanical proper- ties to be optimised and commercial applications developed. Chemical composition, crystallography, microstructure and the concentration of point defects are all factors that have a crucial bearing on superconduc- tivity. Progress in fabrication and thermal processing would elevate these high temperature superconducting oxides from laboratory curiosities to a position where widespread commercial use could be envisaged. Much has already been published on the use of the platinum group metals with the new superconductors. This article has been compiled from a search of the literature and indicates some of the applications that are, or could be, of commercial significance. -
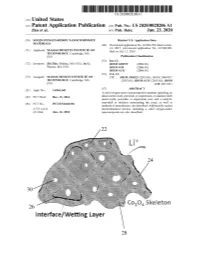
Co , 04 Skeleton Interface / Wetting Layer
US 20200028206A1 IN (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No .: US 2020/0028206 A1 Zhu et al. (43 ) Pub . Date : Jan. 23, 2020 (54 ) SOLID OXYGEN -REDOX NANOCOMPOSITE Related U.S. Application Data MATERIALS (60 ) Provisional application No. 62 / 266,783 , filed on Dec. ( 71) Applicant: MASSACHUSETTS INSTITUTE OF 14 , 2015 , provisional application No. 62 / 360,909 , TECHNOLOGY, Cambridge , MA filed on Jul. 11, 2016 . (US ) Publication Classification (51 ) Int. Ci. (72 ) Inventors : Zhi Zhu , Malden , MA (US ) ; Ju Li, HOIM 10/0525 (2006.01 ) Weston , MA (US ) HOIM 4/38 ( 2006.01) HOIM 4/131 (2006.01 ) (52 ) U.S. CI. ( 73 ) Assignee : MASSACHUSETS INSTITUTE OF CPC .. HOIM 10/0525 ( 2013.01 ) ; HOLM 2004/027 TECHNOLOGY, Cambridge , MA ( 2013.01 ); HOTM 4/131 ( 2013.01 ) ; HOLM (US ) 4/38 ( 2013.01 ) (57 ) ABSTRACT (21 ) Appl. No .: 16 /061,441 A solid oxygen - redox nanocomposite material including an alkali metal oxide, peroxide, or superoxide , or alkaline earth ( 22 ) PCT Filed : Dec. 13 , 2016 metal oxide, peroxide , or superoxide core , and a catalytic nanoshell or skeleton surrounding the cores, as well as ( 86 ) PCT No .: PCT/ US16 /66338 methods of manufacture, are described . Additionally , sealed $ 371 ( c ) ( 1 ) , electrochemical devices including a solid oxygen -redox ( 2 ) Date : Jun . 12 , 2018 nanocomposite are also described . 22 COM 24 30 26 Co , 04 Skeleton Interface /Wetting Layer 28 Patent Application Publication Jan. 23 , 2020 Sheet 1 of 31 US 2020/0028206 A1 22 weeping 24 30 26 C0,0 Skeleton Interface /Wetting Layer 28 Fig . 1 Patent Application Publication Jan. -

Alkylene Oxide Catalysts Having Enhanced Activity And/Or Stability
Europaisches Patentamt J European Patent Office Publication number: 0 393 785 Office europeen des brevets A1 EUROPEAN PATENT APPLICATION © Application number: 90200945.5 © int. el.": C07D 301/10, B01J 23/68 © Date of filing: 17.04.90 © Priority: 18.04.89 US 340390 © Applicant: UNION CARBIDE CHEMICALS AND 18.04.89 US 340242 PLASTICS COMPANY INC. (a New York 03.04.90 US 502187 corporation) 39 Old Ridgebury Road © Date of publication of application: Danbury Connecticut 0681 7-0001 (US) 24.10.90 Bulletin 90/43 © Inventor: Chou, Pen-Yuan © Designated Contracting States: 1331 Wilkie Drive AT BE CH DE DK ES FR GB GR IT LI LU NL SE Charleston, West Virginia 2531 4(US) Inventor: Bhasin, Madan Mohan 9 Carriage Road Charleston, West Virginia 25314(US) Inventor: Soo, Hwaili 1144 Emerald Road Charleston, West Virginia 2531 4(US) Inventor: Thorsteinson, Erlind Magnus 1112 Highland Road Charleston, West Virginia 25302(US) @ Representative: Smulders, Theodorus A.H.J., Ir. et al Vereenigde Octrooibureaux Nieuwe Parklaan 107 NL-2587 BP 's-Gravenhage(NL) © Alkylene oxide catalysts having enhanced activity and/or stability. © Catalysts for the production of alkylene oxide by the epoxidation of alkene with oxygen comprise a silver impregnated support containing a sufficient amount of manganese component to enhance at least one of activity and/or efficiency and/or stability as compared to a similar catalyst which does not contain manganese .component. in oo CO en a. LJJ Xerox Copy Centre EP 0 393 785 A1 ALKYLENE OXIDE CATALYSTS HAVING ENHANCED ACTIVITY AND/OR STABILITY This patent application is a continuation-in-part of U.S. -

University of Groningen Magnetic Order from Molecular Oxygen
University of Groningen Magnetic order from molecular oxygen anions Riyadi, Syarif IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2012 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Riyadi, S. (2012). Magnetic order from molecular oxygen anions. s.n. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 30-09-2021 Chapter 6 Thin Film Rubidium Oxides and Investigation of Cesium and Potassium Oxides 6.1 Introduction Among the alkali metal superoxides, the use of KO2 in breathing apparatus has been well established and widely applied. -
(M2O) Superosides (MO2) and Ozonides (MO3); (M=Li, Na, K) Shiba Subedi*, Jeevan Jyoti Nakarmi
Proceedings International Journal of Nanomaterials, eertechz Nanotechnology and Nanomedicine Special Issue: NCNN-2014 (National Conference on Nanoscience and Nanotechnology - 2014) First Principles Study of Th ermal and Vibrationalfrequencies and Bonding Analysis in Oxides (M2O) Superosides (MO2) and Ozonides (MO3); (M=Li, Na, K) Shiba Subedi*, Jeevan Jyoti Nakarmi Central Department of Physics, Tribhuvan University Kathmandu Nepal, E-mail: [email protected] www.peertechz.com First-principles calculations in order to study the stability of oxides, superoxide and ozonide of alkali metals (Li, Na, K) has been performed by HF cluster procedure implemented by Gaussian 09 sets of programs with the choice of basis set 6-31G*. The correlation effects in the calculations have been accounted by applying Moller-Plesset second order perturbation approximations (MP2). Our calculation shows that when increasing the bond length between atoms of molecules, the binding energy of the corresponding system decreases, i.e. the stability of the studied system is inversely proportional to the bond length between the atoms of the corresponding system. Quantum Theory of Atoms In Molecule (QTAIM) approach has been adopted for bonding analysis for studied systems and this study shows that the bonding in all studied systems are of closed shell type at all. Our study for the variation of the real space function with bond length fi nds that with increase in bond length the real space function values at BCP (ρ and 2ρ) found to be decrease, which are in close agreement with the previously reported calculations. The electrostatic potential within the molecular surface of the studied systems has been systematically analysed. -

Alkali Metal Corrosion of Alumina : Thermodynamics, Phase Diagrams and Testing
Alkali metal corrosion of alumina : thermodynamics, phase diagrams and testing Citation for published version (APA): Hoek, van, J. A. M. (1990). Alkali metal corrosion of alumina : thermodynamics, phase diagrams and testing. Technische Universiteit Eindhoven. https://doi.org/10.6100/IR338654 DOI: 10.6100/IR338654 Document status and date: Published: 01/01/1990 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

United States Patent Office
- 3,030,284 United States Patent Office Patented Apr. 17, 1962 2 e.g., sodium, potassium or lithium oxide. The propor 3,030,284 tion of boric oxide to alkali metal oxide should be such ELECTROLYTIC PRODUCTION OF that the boric oxide is present in a concentration from ELEMENTAL BORON David R. Stern, Fullerton, Calif., assignor to American at least 3% in excess of that required for an alkali metal Potash & Chemical Corporation, a corporation of borate of the formula MO2B2O3, where M is an alkali Delaware metal, up to a total BO3 content of about 95%. Thus, No Drawing. Fied Nov. 3, 1960, Ser. No. 66,902 the minimum boric oxide content for a given alkali metal 4 Claims. (CI. 204-60) borate will be about 3% greater than the value given in the table below, the excess B2O3 being derived by the This is a continuation-in-part of application Serial No. 0 addition thereof to the alkali metal borate. For example, 800,089, filed March 18, 1959, in turn a continuation-in with sodium borate, the BO content of the bath is from part of application Serial No. 574,605, filed March 29, about 73% to about 95% on the total weight of alkali 1956, both now abandoned. metal oxide-boric oxide mixture. The upper limit with This invention relates to the production of highly puri regard to boric oxide is set by the high temperature re fied elemental boron by electrolysis, and more particularly quired to electrolyze the mixture and the difficulty in pro to a process of the foregoing type which may be carried viding a suitable container for the mixture, which is out more or less continuously for extended time periods. -
High Pressure X-Ray Diffraction Study of Sodium Oxide (Na2o): Observations of Amorphization and Equation of State Measurements to 15.9 Gpa
Journal of Alloys and Compounds 823 (2020) 153793 Contents lists available at ScienceDirect Journal of Alloys and Compounds journal homepage: http://www.elsevier.com/locate/jalcom High pressure X-ray diffraction study of sodium oxide (Na2O): Observations of amorphization and equation of state measurements to 15.9 GPa Xiaoxin Wu a, b, Yan Zhang a, b, Junkai Zhang a, b, Ran Liu c, Jinghai Yang a, b, Bin Yang d, * Hongxin Xu a, b, Yanz Ma a, b, a Key Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Changchun, 130103, China b College of Physics, Jilin Normal University, Siping, 136000, China c State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun, 130012, China d Center for High Pressure Science and Technology Advanced Research (HPSTAR), Changchun, 130012, China article info abstract Article history: We have investigated the high-pressure behavior of sodium oxide (Na2O) up to 30 GPa by synchrotron Received 31 August 2019 angle-dispersive powder X-ray diffraction in a diamond anvil cell at room temperature. Between 15.9 and Received in revised form 17.3 GPa crystalline Na2O transforms to an amorphous state, and on decompression the amorphous 8 January 2020 structure recrystallizes back between 4.9 and 10.3 GPa. The zero-pressure bulk modulus of Na Ois Accepted 9 January 2020 2 obtained in experimental observations for the first time. The pressure-volume data is fitted according to Available online 11 January 2020 the third-order Birch-Murnaghan equation of state and yields four bulk modulus values by setting the V0 or B ’ either as a constant or variable. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0230659 A1 Hampden-Smith Et Al
US 2005O230659A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0230659 A1 Hampden-Smith et al. (43) Pub. Date: Oct. 20, 2005 (54) PARTICULATE ABSORBENT MATERIALS Related U.S. Application Data AND METHODS FOR MAKING SAME (63) Continuation-in-part of application No. 10/723,424, (76) Inventors: Mark J. Hampden-Smith, filed on Nov. 26, 2003. Albuquerque, NM (US); Paolina Atanassova, Albuquerque, NM (US); (60) Provisional application No. 60/525,462, filed on Nov. Jian-Ping Shen, Albuquerque, NM 26, 2003. Provisional application No. 60/525,467, (US); James Brewster, Rio Rancho, filed on Nov. 26, 2003. NM (US); Paul Napolitano, Publication Classification Albuquerque, NM (US); Agathagelos Kyrlidis, Malden, MA (US) (51) Int. Cl." ....................................................... C09K 3/00 (52) U.S. Cl. .............................................................. 252/189 Correspondence Address: (57) ABSTRACT MARSH, FISCHMANN & BREYFOGLE LLP Solid absorbent materials that are useful for absorption of 3151 SOUTH WAUGHN WAY chemical Species from a fluid, Such as a gas Stream or a SUTE 411 liquid Stream. The absorbent materials are formed by Spray AURORA, CO 80014 (US) processing and posses a well-defined chemical composition and microstructure. The absorbent materials can have a high absorption capacity for a chemical Species Such as H2S, (21) Appl. No.: 10/996,672 CO, NO, and H and have a high recylability, such that the chemical Species can be absorbed and desorbed over a large (22) Filed: Nov. 24, 2004 number of cycles. Increasing Temperature and Time old Liquid Phase Solid State Ca(NO), -- Pore formers Patent Application Publication Oct. 20, 2005 Sheet 1 of 57 US 2005/0230659 A1 Qu?LpueQunqeJ?duºLºu?seQJOUI V o SIQUILIOJQJOGI Patent Application Publication Oct.