Molecular Analysis of the Aldolase B Gene in Patients with Hereditary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Open Full Page
Research Article Glutathione Transferase P Plays a Critical Role in the Development of Lung Carcinogenesis following Exposure to Tobacco-Related Carcinogens and Urethane Kenneth J. Ritchie,1 Colin J. Henderson,1 Xiu Jun Wang,1 Olga Vassieva,1 Dianne Carrie,1 Peter B. Farmer,2 Margaret Gaskell,2 Kevin Park,3 and C. Roland Wolf1 1Cancer Research UK Molecular Pharmacology Unit, Biomedical Research Centre, Ninewells Hospital and Medical School, Dundee, United Kingdom; 2Cancer Biomarkers and Prevention Group, Biocentre, University of Leicester, Leicester, United Kingdom;and 3Department of Pharmacology and Therapeutics, University of Liverpool, Liverpool, United Kingdom Abstract family of dimeric enzymes (EC 2.5.1.18;GST a, A, k, u, j, ~, n, and N) Human cancer is controlled by a complex interaction between identified on the basis of their amino acid sequence and substrate genetic and environmental factors. Such environmental specificity (4). GSTs are regarded as being important detoxification factors are well defined for smoking-induced lung cancer; enzymes due to their capacity to catalyze the addition of reduced however, the roles of specific genes have still to be elucidated. glutathione (GSH) to reactive electrophiles produced by cyto- Glutathione transferase P (GSTP) catalyzes the detoxification chrome P450 metabolism. As a consequence, there has been a of electrophilic diol epoxides produced by the metabolism of significant interest in elucidating the relationship between GSTP polycyclic aromatic hydrocarbons such as benzo[a]pyrene function and resistance to cancer chemotherapeutic agents and the development of cancer (5–7). In a genetic approach to study GST (BaP), a common constituent of tobacco smoke. Activity- altering polymorphisms in Gstp have therefore been speculated functions, we have generated mice nulled at the Gstp gene locus to be potential risk modifiers in lung cancer development. -

Isolation and Characterization of a Mutant Liver Aldolase in Adult Hereditary Fructose Intolerance
Isolation and characterization of a mutant liver aldolase in adult hereditary fructose intolerance. Identification of the enzyme variant by radioassay in tissue biopsy specimens Timothy M. Cox, … , Michael Camilleri, Arthur H. Burghes J Clin Invest. 1983;72(1):201-213. https://doi.org/10.1172/JCI110958. Hereditary fructose intolerance (HFI) is a metabolic disorder caused by enzymic deficiency of aldolase B, a genetically distinct cytosolic isoenzyme expressed exclusively in liver, kidney, and intestine. The molecular basis of this enzyme defect has been investigated in three affected individuals from a nonconsanguineous kindred, in whom fructose-l- phosphate aldolase activities in liver or intestinal biopsy samples were reduced to 2-6% of mean control values. To identify a putative enzyme mutant in tissue extracts, aldolase B was purified from human liver by affinity chromatography and monospecific antibodies were prepared from antiserum raised in sheep. Immunodiffusion gels showed a single precipitin line common to pure enzyme and extracts of normal liver and intestine, but no reaction with extracts of brain, muscle, or HFI liver. However, weak positive staining for aldolase in hepatocyte and enterocyte cytosol was demonstrated by indirect immunofluorescence of HFI tissues. This was abolished by pretreatment with pure enzyme protein. Accordingly, a specific radioimmunoassay (detection limit 7.5 ng) was established to quantify immunoreactive aldolase B in human biopsy specimens. Extracts of tissue from affected patients gave 10-25% immunoreactive enzyme in control samples; immunoreactive aldolase in intestinal extracts from four heterozygotes was reduced (to 55%) when compared with seven samples from normal control subjects (P < 0.05). In extracts of HFI tissues, there was a sevenfold reduction in apparent absolute specific […] Find the latest version: https://jci.me/110958/pdf Isolation and Characterization of a Mutant Liver Aldolase in Adult Hereditary Fructose Intolerance IDENTIFICATION OF THE ENZYME VARIANT BY RADIOASSAY IN TISSUE BIOPSY SPECIMENS TIMOTHY M. -

Antenatal Diagnosis of Inborn Errors Ofmetabolism
816 ArchivesofDiseaseinChildhood 1991;66: 816-822 CURRENT PRACTICE Arch Dis Child: first published as 10.1136/adc.66.7_Spec_No.816 on 1 July 1991. Downloaded from Antenatal diagnosis of inborn errors of metabolism M A Cleary, J E Wraith The introduction of experimental treatment for Sample requirement and techniques used in lysosomal storage disorders and the increasing prenatal diagnosis understanding of the molecular defects behind By far the majority of antenatal diagnoses are many inborn errors have overshadowed the fact performed on samples obtained by either that for many affected families the best that can amniocentesis or chorion villus biopsy. For be offered is a rapid, accurate prenatal diag- some disorders, however, the defect is not nostic service. Many conditions remain at best detectable in this material and more invasive only partially treatable and as a consequence the methods have been applied to obtain a diagnos- majority of parents seek antenatal diagnosis in tic sample. subsequent pregnancies, particularly for those disorders resulting in a poor prognosis in terms of either life expectancy or normal neurological FETAL LIVER BIOPSY development. Fetal liver biopsy has been performed to The majority of inborn errors result from a diagnose ornithine carbamoyl transferase defi- specific enzyme deficiency, but in some the ciency and primary hyperoxaluria type 1. primary defect is in a transport system or Glucose-6-phosphatase deficiency (glycogen enzyme cofactor. In some conditions the storage disease type I) could also be detected by biochemical defect is limited to specific tissues this method. The technique, however, is inva- only and this serves to restrict the material avail- sive and can be performed by only a few highly able for antenatal diagnosis for these disorders. -
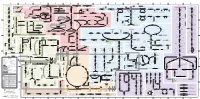
Fullsubwaymap221.Pdf
A B C D E F G H I J K L M Aldose reductase Sorbitol dehydrogenase Cholesterol synthesis Glycogen and galactose metabolism (branch point only) (debranching) glucose sorbitol fructose Aromatic amino acid metabolism Purine synthesis Pyrimidine synthesis glycogen (n+1) phenylacetate triiodothyronine (T3) dopaquinone melanin + + thyroxine (T4) (multiple steps) (many cycles) P 4' NADPH NADP NAD NADH acetyl-CoA x2 i Debranching enzyme 3' - α-1,4 bonds Aromatic amino acid 5-phosphoribosyl pyrophosphate (PRPP) HCO B 2' P 3 (branching) Glycogen 6 i (multiple steps) O H O (multiple steps) Tyrosinase O H O 1' 2 2 2 2 B 1 2 3 4 5 6 7 8 9 10 Pentose phosphate pathway Phenylalanine Tyrosine decarboxylase 6 Dopamine B Thiolase phosphorylase -5 -4 -3 -2 -1 0 1 2 3 4 Transaminase 6 glutamine 2 ATP (many cycles) Glycogen Glucose-6-phosphatase Dehydrogenase hydroxylase hydroxylase (DOPA decarboxylase) β-hydroxylase Methyltransferase 10 UDP 4:4 Transferase ATP glutathione (reduced) H O phenyllactate phenylpyruvate phenylalanine tyrosine dihydroxy- dopamine Glutamine PRPP CoA synthase Hexokinase or (in endoplasmic reticulum) 2 2 norepinephrine epinephrine glutamine Carbamoyl 9 1' phenylanine amidotransferase (GPAT) glutamate glycogen (n) Glutathione reductase Glutathione peroxidase + phosphate glycogen (n) -5 -4 -3 -2 -1 0 1 2 3 4 2' 3' 4' glucokinase 8 NADP NADPH glutamate α-ketoglutarate CO2 O H O Glycosyl (4:6) UDP-glucose ADP (L-DOPA) 2 2 synthetase II glutathione (oxidized) 2 H O α-ketoglutarate PPi glutamate acetoacetyl-CoA 7 1 Glucose -1,6-Glucosidase -

Teladorsagia Circumcincta 1,6 Bisphosphate Aldolase: Molecular and Biochemical
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201335; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Teladorsagia circumcincta 1,6 bisphosphate aldolase: molecular and biochemical 2 characterisation, structure analysis and recognition by immune hosts 3 4 5 6 7 S. Umaira*, C.L.G. Boucheta, N. Palevicha, J.S. Knighta, H.V. Simpsonb 8 9 10 11 aAgResearch Ltd, Private Bag 11-008, Palmerston North, New Zealand 12 bInstitute of Veterinary, Animal and Biomedical Sciences, Massey University, Private 13 Bag 11-222, Palmerston North, New Zealand 14 15 * Corresponding author 16 Saleh Umair, AgResearch Ltd, Hopkirk Research Institute, Grasslands Research 17 Centre, Tennent Drive, Private Bag 11008, Palmerston North 4442, New Zealand. 18 E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.13.201335; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 ABSTRACT 2 A 1095 bp full length cDNA encoding Teladorsagia circumcincta aldolase 3 (TciALDO) was cloned, expressed in Escherichia coli, the recombinant protein 4 purified and its kinetic properties determined. A phylogenetic tree was constructed 5 using helminth aldolase sequences. -

Supplementary Information
Supplementary Information Table S1. Identification of amyloid-binding proteins in mouse brain homogenates. No. Protein Name Acc. Number a Mol. Mass Score b Peptides Localization Function 1 Carbamoyl-phosphate synthase [ammonia], mitochondrial CPSM_RAT 164,476 1129 15 M 6 2 Pyruvate kinase isozymes M1/M2 KPYM_RAT 57,781 706 7 C, N 1 3 Betaine—Homocysteine S-methyltransferase 1 BHMT1_RAT 44,948 650 2 C 6 4 Adenylate kinase 2, mitochondrial KAD2_RAT 26,363 540 4 M 1 5 δ-1-Pyrroline-5-carboxylate dehydrogenase, mitochondrial AL4A1_RAT 61,830 538 4 M 6 6 Actin, cytoplasmic 1 ACTB_RAT 41,710 524 3 C 2 # 7 Myosin-4 MYH4_RAT 222,741 396 4 C 2 8 3-Ketoacyl-CoA thiolase, mitochondrial THIM_RAT 41,844 350 1 M 7 9 Hydroxyacyl-coenzyme A dehydrogenase, mitochondrial HCDH_RAT 34,426 310 3 M 7 10 Heat shock cognate 71 kDa protein HSP7C_RAT 70,827 305 3 C, N, S, Mel 5 # 11 ATP synthase subunit β, mitochondrial ATPB_RAT 56,318 290 4 M 1 # 12 Calmodulin CALM_RAT 16,827 288 1 C 2 # 13 Fructose-bisphosphate aldolase B ALDOB_RAT 39,593 249 2 C, L, N, ER, CM 1 # 14 α-Enolase ENOA_RAT 47,098 220 2 CM, C, Me 1 # 15 Ornithine carbamoyltransferase, mitochondrial OTC_RAT 39,861 212 5 M 6 16 Myelin basic protein S MBP_RAT 21,489 211 1 CM, Me 5 17 Glutathione S-transferase α-2 GSTA2_RAT 25,543 208 1 C, M, N 5 18 Phosphatidylethanolamine-binding protein 1 PEBP1_RAT 20,788 182 1 C, Me 4 19 Dihydropyrimidinase-related protein 2 DPYL2_RAT 62,239 177 2 C, Me, M 3 # 20 60 kDa Heat shock protein, mitochondrial CH60_RAT 60,917 162 4 M 5 21 Glyceraldehyde-3-phosphate dehydrogenase G3P_RAT 35,787 160 1 C, N 1 # 22 Electron transfer flavoprotein subunit α, mitochondrial ETFA_RAT 34,929 137 3 M 1 23 Fructose-bisphosphate aldolase A ALDOA_RAT 39,327 135 2 Me, M 1 24 ATP synthase subunit α, mitochondrial ATPA_RAT 59,717 131 2 M 1 25 Thioredoxin-dependent peroxide reductase, mitochondrial PRDX3_RAT 28,277 130 1 M 5 26 Elongation factor 1-α 1 EF1A1_RAT 50,082 129 2 N 3 27 Endoplasmin ENPL_RAT 92,713 125 1 ER 5 S2 Table S1. -

ALDOB Gene Aldolase, Fructose-Bisphosphate B
ALDOB gene aldolase, fructose-bisphosphate B Normal Function The ALDOB gene provides instructions for making the aldolase B enzyme. This enzyme is one of a group of three aldolase enzymes that are responsible for breaking down certain molecules in cells throughout the body. Four identical aldolase B enzymes need to be attached (bound) to each other in a four-enzyme unit called a tetramer to work. Aldolase B is found primarily in the liver, but it is also present at lower levels in kidney and intestinal cells. Aldolase B is involved in the breakdown (metabolism) of the simple sugar fructose, which is found mostly in fruits and is used in the body for energy. Aldolase B is responsible for the second step in the metabolism of fructose, which breaks down the molecule fructose-1-phosphate into glyceraldehyde and dihydroxyacetone phosphate. To a lesser degree, aldolase B is also involved in the breakdown of the simple sugar glucose. Health Conditions Related to Genetic Changes Hereditary fructose intolerance More than 50 mutations in the ALDOB gene have been found to cause hereditary fructose intolerance, a condition characterized by nausea and intestinal discomfort following ingestion of foods containing fructose. Most of these mutations replace single protein building blocks (amino acids) in the aldolase B enzyme and result in the production of an enzyme with reduced function. A mutation found in approximately half of people with hereditary fructose intolerance replaces the amino acid alanine with the amino acid proline at position 149 in the enzyme (written as Ala149Pro or A149P). This mutation alters the 3-dimensional shape of the enzyme. -

Introduction the Terms in the Subject Index for Volume 81, January
Introduction The terms in the Subject Index for Volume 81, January-December 1984, of the PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES USA (Biological Sci- ences) were chosen from titles, key terms, and abstracts of articles. The index terms are alphabetized by computer; numbers, conformational prefixes, and hy- phenated Greek letters are disregarded in alphabetization. 8041 8042 Biological Sciences Subject Index Proc. Natl. Acad. Sci. USA 81 (1984) A* protein Different channel properties of Torpedo acetylcholine receptor monomers Cleavage of single-stranded DNA by the <X174 A* protein: The A*- and dimers reconstituted in planar membranes, 6222 single-stranded DNA covalent linkage, 4285 Phosphorylation of the nicotinic acetylcholine receptor by an endogenous Abelson murine leukemia virus tyrosine-specific protein kinase, 6968 Transformation-associated proteins in murine B-cell lymphomas that are Purification of the muscarinic acetylcholine receptor from porcine atria distinct from Abelson virus gene products, 4434 (Correction), 7258 Abortive infection Acetylcholine receptor-specific suppressive T-cell factor from a Virus-plasmid interactions: Mutants of bacteriophage T3 that abortively retrovirally transformed T-cell line, 7569 infect plasmid F-containing (F+) strains of Escherichia coli, 1465 Isolation and characterization of a cDNA clone for the complete protein Cytomegalovirus infects human lymphocytes and monocytes: Virus coding region of the 8 subunit of the mouse acetylcholine expression is restricted to immediate-early gene products, -

Transcriptional Control of Genes That Regulate Glycolysis and Gluconeogenesis in Adult Liver Frederic P
Biochem. J. (1994) 303, 1-14 (Printed in Great Britain) 1 REVIEW ARTICLE Transcriptional control of genes that regulate glycolysis and gluconeogenesis in adult liver Frederic P. LEMAIGRE and Guy G. ROUSSEAU* Hormone and Metabolic Research Unit, University of Louvain Medical School and International Institute of Cellular and Molecular Pathology, 75 Avenue Hippocrate, B-1200 Brussels, Belgium INTRODUCTION listed in Table 1. The ubiquitous factors that control the genes discussed here are listed in Table 2. These genes code for Glycolysis is the metabolic pathway through which glucose is glucokinase, 6-phosphofructo-2-kinase (PFK-2)/fructose-2,6- converted into pyruvate, with a net yield of 2 mol of ATP and bisphosphatase (FBPase-2), pyruvate kinase and phosphoenol- NADH per mol of glucose used. In liver, where the glycolytic pyruvate carboxykinase (PEPCK), namely enzymes that control flux is low except when glucose concentrations are high or key steps, and for aldolase and glyceraldehyde-3-phosphate during anoxia, the main function of glycolysis is to provide dehydrogenase (GAPDH), which are enzymes controlling re- substrates for anabolic processes. While glycolysis occurs in versible steps. every tissue, gluconeogenesis is specific for the liver, the kidney In the present paper, we will review the data pertaining to the and the small intestine. Gluconeogenesis supplies glucose to the transcriptional control of these genes in liver. We will describe body and provides a way to dispose of amino acids and lactate first the glycolytic enzymes and then the gluconeogenic enzyme produced by erythrocytes and during muscle contraction. It is PEPCK. Aldolase B and GAPDH are discussed with the also a means of disposal for glycerol produced during lipolysis. -

Zinc Metalloenzymes in Plants
ZINC METALLOENZYMES IN PLANTS JORGE CASTILLO-GONZÁLEZ, DÁMARIS OJEDA-BARRIOS, ADRIANA HERNÁNDEZ-RODRÍGUEZ, ANA CECILIA GONZÁLEZ-FRANCO, LORETO ROBLES-HERNÁNDEZ and GUSTAVO ROGELIO LÓPEZ-OCHOA SUMMARY Zinc is an essential plant micronutrient and soil availability protein synthesis. Plants also require zinc for the synthesis of is of great importance in many crops. In plants, zinc is neither tryptophan, a key amino acid in the synthesis of the auxin in- oxidized nor reduced; instead, the significance of zinc stems doleacetic acid. Therefore, zinc also operates in the control of from its physiochemical properties as a divalent cation. Many plant development through its indirect action on auxins. Zinc enzymes include zinc as a cofactor, like the alcohol dehydro- deficiency affects the catalytic activity of all the above enzymes genase (EC 1.1.1.1), superoxide dismutase (EC 1.15.1.1), carbo- and, thus, the metabolic pathways in which they are involved. nic anhydrase (EC 4.2.1.1) and RNA polymerase (EC 2.7.7.6). In The aim of this paper is to analyze the function of Zn in some these cases, it is indirectly evident that zinc deficiency inhibits of metalloproteins involved in plant metabolism. n contrast to Fe, Mn, Cu Zn can give support and stability to an (Hong et al., 2007). Changes in plant me- and Mo, Zn is a transition enzyme by activating it. Higher plants tabolism induced by Zn deficiency include element that is not subject have a number of enzymes containing effects on carbohydrates, proteins, auxins to valence change and, thus, Zn, including alcohol dehydrogenase, car- and damage to membrane integrity. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Exogenous Spermidine Enhances Expression of Calvin Cycle Genes and Photosynthetic Efficiency in Sweet Sorghum Seedlings Under Salt Stress
DOI: 10.32615/bp.2019.046 BIOLOGIA PLANTARUM 63: 511-518, 2019 This is an open access article distributed under the terms of the Creative Commons BY-NC-ND Licence Exogenous spermidine enhances expression of Calvin cycle genes and photosynthetic efficiency in sweet sorghum seedlings under salt stress A.I. EL SAYED1*, M.A.M. EL-HAMAHMY2, M.S. RAFUDEEN3, and M.K.H. EBRAHIM4,5 Biochemistry Department, Faculty of Agriculture, Zagazig University, 44519 Zagazig, Egypt1 Department of Agricultural Botany, Faculty of Agriculture, Suez Canal University, 41522 Ismailia, Egypt2 Department of Molecular and Cell Biology, University of Cape Town, Private Bag, 7701 Rondebosch, South Africa3 Biology Department, Faculty of applied sciences, Umm Al- Qura University, postal code? Makkah Al- Mukarramah, KSA4 Botany Department, Faculty of Science, Tanta University, 31257 Tanta, Egypt 5 Abstract Salinity adversely affects plants resulting in disruption to plant growth and physiology. Previously, it has been shown that these negative effects can be alleviated by various exogenous polyamines. However, the role of spermidine (Spd) in conferring salinity tolerance in sorghum is not well documented. The effect of exogenous Spd on the responses of sweet sorghum (Sorghum bicolor L.) seedlings to salt stress (150 mM NaCl) was investigated by measuring photosynthetic carbon assimilation, Calvin cycle enzyme activities, and the the expression of respective genes. Application of 0.25 mM Spd alleviated the negative effects of salt stress on efficiency of photosystem II and CO2 assimilation and increased the activities of ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) and aldolase. Salt stress significantly lowered the transcriptions of genes encoding Rubisco large subunit, Rubisco small subunit, 3-phosphoglyceric acid kinase, glyceraldehyde-3-phosphate dehydrogenase, triose-3-phosphate isomerase, fructose-1,6-bisphosphate aldolase, fructose- 1,6-bisphosphate phosphatase, and sedoheptulose-1,7-bisphosphatase.