Mismatch Repair: from Preserving Genome Stability to Enabling Mutation Studies in Real-Time Single Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deficiency in the DNA Repair Protein ERCC1 Triggers a Link Between Senescence and Apoptosis in Human Fibroblasts and Mouse Skin
Lawrence Berkeley National Laboratory Recent Work Title Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Permalink https://escholarship.org/uc/item/73j1s4d1 Journal Aging cell, 19(3) ISSN 1474-9718 Authors Kim, Dong Eun Dollé, Martijn ET Vermeij, Wilbert P et al. Publication Date 2020-03-01 DOI 10.1111/acel.13072 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Received: 10 June 2019 | Revised: 7 October 2019 | Accepted: 30 October 2019 DOI: 10.1111/acel.13072 ORIGINAL ARTICLE Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin Dong Eun Kim1 | Martijn E. T. Dollé2 | Wilbert P. Vermeij3,4 | Akos Gyenis5 | Katharina Vogel5 | Jan H. J. Hoeijmakers3,4,5 | Christopher D. Wiley1 | Albert R. Davalos1 | Paul Hasty6 | Pierre-Yves Desprez1 | Judith Campisi1,7 1Buck Institute for Research on Aging, Novato, CA, USA Abstract 2Centre for Health Protection Research, ERCC1 (excision repair cross complementing-group 1) is a mammalian endonuclease National Institute of Public Health and that incises the damaged strand of DNA during nucleotide excision repair and inter- the Environment (RIVM), Bilthoven, The −/Δ Netherlands strand cross-link repair. Ercc1 mice, carrying one null and one hypomorphic Ercc1 3Department of Molecular Genetics, allele, have been widely used to study aging due to accelerated aging phenotypes Erasmus University Medical Center, −/Δ Rotterdam, The Netherlands in numerous organs and their shortened lifespan. Ercc1 mice display combined 4Princess Máxima Center for Pediatric features of human progeroid and cancer-prone syndromes. -

Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases
cells Review Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases Paulina Prorok 1 , Inga R. Grin 2,3, Bakhyt T. Matkarimov 4, Alexander A. Ishchenko 5 , Jacques Laval 5, Dmitry O. Zharkov 2,3,* and Murat Saparbaev 5,* 1 Department of Biology, Technical University of Darmstadt, 64287 Darmstadt, Germany; [email protected] 2 SB RAS Institute of Chemical Biology and Fundamental Medicine, 8 Lavrentieva Ave., 630090 Novosibirsk, Russia; [email protected] 3 Center for Advanced Biomedical Research, Department of Natural Sciences, Novosibirsk State University, 2 Pirogova St., 630090 Novosibirsk, Russia 4 National Laboratory Astana, Nazarbayev University, Nur-Sultan 010000, Kazakhstan; [email protected] 5 Groupe «Mechanisms of DNA Repair and Carcinogenesis», Equipe Labellisée LIGUE 2016, CNRS UMR9019, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94805 Villejuif, France; [email protected] (A.A.I.); [email protected] (J.L.) * Correspondence: [email protected] (D.O.Z.); [email protected] (M.S.); Tel.: +7-(383)-3635187 (D.O.Z.); +33-(1)-42115404 (M.S.) Abstract: It was proposed that the last universal common ancestor (LUCA) evolved under high temperatures in an oxygen-free environment, similar to those found in deep-sea vents and on volcanic slopes. Therefore, spontaneous DNA decay, such as base loss and cytosine deamination, was the Citation: Prorok, P.; Grin, I.R.; major factor affecting LUCA’s genome integrity. Cosmic radiation due to Earth’s weak magnetic field Matkarimov, B.T.; Ishchenko, A.A.; and alkylating metabolic radicals added to these threats. -

Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA
Mechanisms in E. coli and Human Mismatch Repair Nobel Lecture, December 8, 2015 by Paul Modrich Howard Hughes Medical Institute and Department of Biochemistry, Duke University Medical Center, Durham, North Carolina, USA. he idea that mismatched base pairs occur in cells and that such lesions trig- T ger their own repair was suggested 50 years ago by Robin Holliday in the context of genetic recombination [1]. Breakage and rejoining of DNA helices was known to occur during this process [2], with precision of rejoining attributed to formation of a heteroduplex joint, a region of helix where the two strands are derived from the diferent recombining partners. Holliday pointed out that if this heteroduplex region should span a genetic diference between the two DNAs, then it will contain one or more mismatched base pairs. He invoked processing of such mismatches to explain the recombination-associated phenomenon of gene conversion [1], noting that “If there are enzymes which can repair points of damage in DNA, it would seem possible that the same enzymes could recognize the abnormality of base pairing, and by exchange reactions rectify this.” Direct evidence that mismatches provoke a repair reaction was provided by bacterial transformation experiments [3–5], and our interest in this efect was prompted by the Escherichia coli (E. coli) work done in Matt Meselson’s lab at Harvard. Using artifcially constructed heteroduplex DNAs containing multiple mismatched base pairs, Wagner and Meselson [6] demonstrated that mismatches elicit a repair reaction upon introduction into the E. coli cell. Tey also showed that closely spaced mismatches, mismatches separated by a 1000 base pairs or so, are usually repaired on the same DNA strand. -

DNA Repair with Its Consequences (E.G
Cell Science at a Glance 515 DNA repair with its consequences (e.g. tolerance and pathways each require a number of apoptosis) as well as direct correction of proteins. By contrast, O-alkylated bases, Oliver Fleck* and Olaf Nielsen* the damage by DNA repair mechanisms, such as O6-methylguanine can be Department of Genetics, Institute of Molecular which may require activation of repaired by the action of a single protein, Biology, University of Copenhagen, Øster checkpoint pathways. There are various O6-methylguanine-DNA Farimagsgade 2A, DK-1353 Copenhagen K, Denmark forms of DNA damage, such as base methyltransferase (MGMT). MGMT *Authors for correspondence (e-mail: modifications, strand breaks, crosslinks removes the alkyl group in a suicide fl[email protected]; [email protected]) and mismatches. There are also reaction by transfer to one of its cysteine numerous DNA repair pathways. Each residues. Photolyases are able to split Journal of Cell Science 117, 515-517 repair pathway is directed to specific Published by The Company of Biologists 2004 covalent bonds of pyrimidine dimers doi:10.1242/jcs.00952 types of damage, and a given type of produced by UV radiation. They bind to damage can be targeted by several a UV lesion in a light-independent Organisms are permanently exposed to pathways. Major DNA repair pathways process, but require light (350-450 nm) endogenous and exogenous agents that are mismatch repair (MMR), nucleotide as an energy source for repair. Another damage DNA. If not repaired, such excision repair (NER), base excision NER-independent pathway that can damage can result in mutations, diseases repair (BER), homologous recombi- remove UV-induced damage, UVER, is and cell death. -

Error-Prone DNA Repair As Cancer's Achilles' Heel
cancers Review Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel Daniele Caracciolo, Caterina Riillo , Maria Teresa Di Martino , Pierosandro Tagliaferri and Pierfrancesco Tassone * Department of Experimental and Clinical Medicine, Magna Græcia University, Campus Salvatore Venuta, 88100 Catanzaro, Italy; [email protected] (D.C.); [email protected] (C.R.); [email protected] (M.T.D.M.); [email protected] (P.T.) * Correspondence: [email protected] Simple Summary: Cancer onset and progression lead to a high rate of DNA damage, due to replicative and metabolic stress. To survive in this dangerous condition, cancer cells switch the DNA repair machinery from faithful systems to error-prone pathways, strongly increasing the mutational rate that, in turn, supports the disease progression and drug resistance. Although DNA repair de-regulation boosts genomic instability, it represents, at the same time, a critical cancer vulnerability that can be exploited for synthetic lethality-based therapeutic intervention. We here discuss the role of the error-prone DNA repair, named Alternative Non-Homologous End Joining (Alt-NHEJ), as inducer of genomic instability and as a potential therapeutic target. We portray different strategies to drug Alt-NHEJ and discuss future challenges for selecting patients who could benefit from Alt-NHEJ inhibition, with the aim of precision oncology. Abstract: Error-prone DNA repair pathways promote genomic instability which leads to the onset of cancer hallmarks by progressive genetic aberrations in tumor cells. The molecular mechanisms which Citation: Caracciolo, D.; Riillo, C.; Di foster this process remain mostly undefined, and breakthrough advancements are eagerly awaited. Martino, M.T.; Tagliaferri, P.; Tassone, In this context, the alternative non-homologous end joining (Alt-NHEJ) pathway is considered P. -

Clinical Significance of ERCC2, XPC, ERCC5 and XRCC3 Gene Polymorphisms in Diffuse Large B Cell Lymphoma
DOI: 10.14744/ejmi.2020.56831 EJMI 2020;4(3):332–340 Research Article Clinical Significance of ERCC2, XPC, ERCC5 and XRCC3 Gene Polymorphisms in Diffuse Large B Cell Lymphoma Aykut Bahceci,1 Semra Paydas,2 Melek Ergin,3 Gulsah Seydaoglu,4 Gulsum Ucar5 1Department of Medical Oncology, Dr. Ersin Arslan Training and Research Hospital, Gaziantep, Turkey 2Department of Medical Oncology, Cukurova University Faculty of Medicine, Adana, Turkey 3Department of Patology, Cukurova University Faculty of Medicine, Adana, Turkey 4Department of Biostatistics, Cukurova University Faculty of Medicine, Adana, Turkey 5Department of Pediatric Hematology, Cukurova University Faculty of Medicine, Adana, Turkey Abstract Objectives: DNA repair genes protects the genome from DNA damage both of endogenous and exogenous stress fac- tors. Due to DNA repair gene polymorphisms, there are differences in the repair capacity between several cancer types. The aim of this study is to evaluate the association between some of the DNA repair gene polymorphisms and clinical outcome in Diffuse Large B-Cell Lymphoma (DLBCL). Methods: The association between clinical factors including stage at diagnosis, extra-nodal involvement, tumor bur- den, bone marrow involvement, relapse status, disease-free/overall survival times and DNA repair gene polymorphisms including ERCC2 (Lys751Gln), XPC (Gln939Lys), ERCC5 (Asp1104His) and XRCC3 (Thr241Met) in 58 patients with DLBCL. T-Shift Real-Time PCR was used to detect these mutations. Results: The median survival times were 60 months and 109 months in patients with CC genotype and CA/AA geno- type of XPC gene polymorphism, respectively (p=0.017). More interestingly, median survival times were 9 months and 109 months in patients with CC (XPC)/CC (XRCC3) and CA/AA (XPC)/CT/TT (XRCC3) for both XPC and XRCC3 gene polymorphisms, respectively (p=0.004). -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -

DNA Damage and Repair
What is DNA? DNA/RNA: polynucleotide chains Phosphate Base Sugar (2’ OH=ribose, 2’H=deoxyribose) Nucleotide =sugar+phosphate+base DNA is a double helix DNA damage and repair • How is DNA damaged? • How is DNA repaired? • How does the type of damage impact repair? • Accumulated DNA damage=death (by cancer, or old age) • “No one here gets out alive” –Jim Morrison Adduct formation • Nasty chemicals(carcinogens) that adduct to DNA; often to ring Nitrogens in bases – E.g. Alkylating agents: reactive carbon containing chemicals (ethylating agents, methylating agents) Adduct formation • Not always direct exposure: sometimes carcinogen is toxic product of cellular metabolism – Cigarette smoke; benzo-a- pyrene not a big deal…but the break down product is • Groups are bulky, blocks transcription, replication; can interfere with base pairing, and introduce mutation during replication Radiation: UV light • Non-ionizing radiation (UV light from the sun) – Bases absorb energy with peak at 260nM..this is UV – Photoactivates base, causes nasty chemistry – Result is…covalent bonds between adajacent bases, almost always adjacent pyrimidines – Distorts DNA (kink), can block transcription, replication, lead to mutation T T T C Spontaneous Damage: Base loss • Some times bases just fall off (more often than you might think; 10000/genome/generation) • Bases gone, but phosphodiester backbone is still intact • Purines more sensitive than pyrimidines (acid sensitive) • Causes mutation, can lead to strand breaks Spontaneous damage: deamination 1. Converts C to U etc… 2. Altered base has different base pairing rule – e.g. U pairs with A (converts CG bp to UA) 3. Unless repaired results in transition mutation Oxidative stress • Reactive oxygen species (ROS); things that are or give rise to oxygen with an unpaired electron; a free radical • E.g hydroxyl radical H O • ROS produced by…. -

The Consequences of Rad51 Overexpression for Normal and Tumor Cells
dna repair 7 (2008) 686–693 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/dnarepair Mini review The consequences of Rad51 overexpression for normal and tumor cells Hannah L. Klein ∗ Department of Biochemistry, New York University School of Medicine, NYU Medical Center, 550 First Avenue, New York, NY 10016, United States article info abstract Article history: The Rad51 recombinase is an essential factor for homologous recombination and the Received 11 December 2007 repair of DNA double strand breaks, binding transiently to both single stranded and double Accepted 12 December 2007 stranded DNA during the recombination reaction. The use of a homologous recombination Published on line 1 February 2008 mechanism to repair DNA damage is controlled at several levels, including the binding of Rad51 to single stranded DNA to form the Rad51 nucleofilament, which is controlled through Keywords: the action of DNA helicases that can counteract nucleofilament formation. Overexpression Rad51 protein of Rad51 in different organisms and cell types has a wide assortment of consequences, rang- Overexpression of Rad51 ing from increased homologous recombination and increased resistance to DNA damaging Genomic instability agents to disruption of the cell cycle and apoptotic cell death. Rad51 expression is increased Tumor cell drug resistance in p53-negative cells, and since p53 is often mutated in tumor cells, there is a tendency for Homologous recombination Rad51 to be overexpressed in tumor cells, leading to increased resistance to DNA damage Gene targeting and drugs used in chemotherapies. As cells with increased Rad51 levels are more resis- tant to DNA damage, there is a selection for tumor cells to have higher Rad51 levels. -

Recruitment of Mismatch Repair Proteins to the Site of DNA Damage in Human Cells
3146 Research Article Recruitment of mismatch repair proteins to the site of DNA damage in human cells Zehui Hong, Jie Jiang, Kazunari Hashiguchi, Mikiko Hoshi, Li Lan and Akira Yasui* Department of Molecular Genetics, Institute of Development, Aging and Cancer, Tohoku University, Seiryomachi 4-1, Aobaku, Sendai 980-8575, Japan *Author for correspondence (e-mail: [email protected]) Accepted 8 July 2008 Journal of Cell Science 121, 3146-3154 Published by The Company of Biologists 2008 doi:10.1242/jcs.026393 Summary Mismatch repair (MMR) proteins contribute to genome stability the PCNA-binding domain of MSH6. MSH2 is recruited to the by excising DNA mismatches introduced by DNA polymerase. DNA damage site through interactions with either MSH3 or Although MMR proteins are also known to influence cellular MSH6, and is required for recruitment of MLH1 to the damage responses to DNA damage, how MMR proteins respond to DNA site. We found, furthermore, that MutSβ is also recruited to damage within the cell remains unknown. Here, we show that UV-irradiated sites in nucleotide-excision-repair- and PCNA- MMR proteins are recruited immediately to the sites of various dependent manners. Thus, MMR and its proteins function not types of DNA damage in human cells. MMR proteins are only in replication but also in DNA repair. recruited to single-strand breaks in a poly(ADP-ribose)- dependent manner as well as to double-strand breaks. Using Supplementary material available online at mutant cells, RNA interference and expression of fluorescence- http://jcs.biologists.org/cgi/content/full/121/19/3146/DC1 -
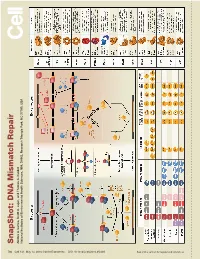
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Involvement of Nucleotide Excision and Mismatch Repair Mechanisms in Double Strand Break Repair
250 Current Genomics, 2009, 10, 250-258 Involvement of Nucleotide Excision and Mismatch Repair Mechanisms in Double Strand Break Repair Ye Zhang1,2,*, Larry H. Rohde2 and Honglu Wu1 1NASA Johnson Space Center, Houston, Texas 77058 and 2University of Houston at Clear Lake, Houston, Texas 77058, USA Abstract: Living organisms are constantly threatened by environmental DNA-damaging agents, including UV and ioniz- ing radiation (IR). Repair of various forms of DNA damage caused by IR is normally thought to follow lesion-specific re- pair pathways with distinct enzymatic machinery. DNA double strand break is one of the most serious kinds of damage induced by IR, which is repaired through double strand break (DSB) repair mechanisms, including homologous recombi- nation (HR) and non-homologous end joining (NHEJ). However, recent studies have presented increasing evidence that various DNA repair pathways are not separated, but well interlinked. It has been suggested that non-DSB repair mecha- nisms, such as Nucleotide Excision Repair (NER), Mismatch Repair (MMR) and cell cycle regulation, are highly involved in DSB repairs. These findings revealed previously unrecognized roles of various non-DSB repair genes and indicated that a successful DSB repair requires both DSB repair mechanisms and non-DSB repair systems. One of our recent studies found that suppressed expression of non-DSB repair genes, such as XPA, RPA and MLH1, influenced the yield of IR- induced micronuclei formation and/or chromosome aberrations, suggesting that these genes are highly involved in DSB repair and DSB-related cell cycle arrest, which reveals new roles for these gene products in the DNA repair network.