Kinetic Interval Measurement: a Tool to Characterize Thermal Reversion Dynamics of Light-Switchable Fluorescent Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evos Cell Imaging Analysis Systems
Imaging, labeling, and detection solutions Microscopy | High-content analysis | Cell counting | Plate reading Minimizing the complexities of cellular analysis Our cellular analysis product portfolio combines the strengths of Invitrogen™ fluorescent reagents and a complete line of versatile analysis instrumentation. Select from a line of heavily peer-referenced platforms to make the discoveries that catalyze advances toward your research goals of tomorrow. Invitrogen™ EVOS™ imaging systems Our comprehensive imaging portfolio includes: • Cell imaging systems • Automated cell counting systems • High-content analysis systems Invitrogen™ Countess™ cell counters • Cell imaging reagents • Microplate readers All of our analysis systems are designed to work together— Thermo Scientific™ CellInsight™ from the initial cell culture check to more complex analyses. high-content analysis systems Discover more about your samples with automated cell counting, long-term live-cell imaging, automated multiwell plate scanning, and phenotypic screening. Thermo Scientific™ Varioskan™ LUX multimode plate reader Contents Microscopy Compact and portable imaging systems 4 EVOS imaging systems at a glance 5 EVOS M7000 Imaging System 6 Live-cell imaging with the Onstage Incubator 8 Image analysis with Celleste software 10 EVOS M5000 Imaging System 12 EVOS FLoid Imaging Station 14 EVOS XL Core Imaging System 15 EVOS vessel holders and stage plates 16 The power of LED illumination 17 EVOS objectives 18 Fluorophore selection guide 20 High-content analysis CellInsight high-content analysis platforms 22 CellInsight CX7 LZR High-Content Analysis Platform 24 CellInsight CX5 High-Content Screening Platform 25 HCS Studio Cell Analysis Software 26 Store Image and Database Management Software 27 Cell counting Countess II Automated Cell Counters 28 Plate reading Microplate readers 30 Microscopy Compact and portable imaging systems Now you can have an easy-to-use cell imaging platform From intimate hands-on demonstrations to presentations of where you want it and when you want it. -

ELISA Plate Reader
applications guide to microplate systems applications guide to microplate systems GETTING THE MOST FROM YOUR MOLECULAR DEVICES MICROPLATE SYSTEMS SALES OFFICES United States Molecular Devices Corp. Tel. 800-635-5577 Fax 408-747-3601 United Kingdom Molecular Devices Ltd. Tel. +44-118-944-8000 Fax +44-118-944-8001 Germany Molecular Devices GMBH Tel. +49-89-9620-2340 Fax +49-89-9620-2345 Japan Nihon Molecular Devices Tel. +06-6399-8211 Fax +06-6399-8212 www.moleculardevices.com ©2002 Molecular Devices Corporation. Printed in U.S.A. #0120-1293A SpectraMax, SoftMax Pro, Vmax and Emax are registered trademarks and VersaMax, Lmax, CatchPoint and Stoplight Red are trademarks of Molecular Devices Corporation. All other trademarks are proprty of their respective companies. complete solutions for signal transduction assays AN EXAMPLE USING THE CATCHPOINT CYCLIC-AMP FLUORESCENT ASSAY KIT AND THE GEMINI XS MICROPLATE READER The Molecular Devices family of products typical applications for Molecular Devices microplate readers offers complete solutions for your signal transduction assays. Our integrated systems γ α β s include readers, washers, software and reagents. GDP αs AC absorbance fluorescence luminescence GTP PRINCIPLE OF CATCHPOINT CYCLIC-AMP ASSAY readers readers readers > Cell lysate is incubated with anti-cAMP assay type SpectraMax® SpectraMax® SpectraMax® VersaMax™ VMax® EMax® Gemini XS LMax™ ATP Plus384 190 340PC384 antibody and cAMP-HRP conjugate ELISA/IMMUNOASSAYS > nucleus Single addition step PROTEIN QUANTITATION cAMP > λEX 530 nm/λEM 590 nm, λCO 570 nm UV (280) Bradford, BCA, Lowry For more information on CatchPoint™ assay NanoOrange™, CBQCA kits, including the complete procedure for this NUCLEIC ACID QUANTITATION assay (MaxLine Application Note #46), visit UV (260) our web site at www.moleculardevices.com. -

Dynapro® Plate Reader™ II User's Guide
DynaPro® Plate Reader™ II User’s Guide M3101 Rev C Copyright © 2014, Wyatt Technology Corporation WYATT TECHNOLOGY Corp., makes no warranties, either express or implied, regarding this instrument, computer software package, its merchantability or its fitness for any particular purpose. The software is provided “as is,” without warranty of any kind. Furthermore, Wyatt Technology does not warrant, guarantee, or make any representations regarding the use, or the results of the use, of the software or written materials in terms of correctness, accuracy, reliability, currentness, or otherwise. The exclusion of implied warranties is not permitted by some states, so the above exclusion may not apply to you. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form by any means, electronic, mechanical, photocopying, recording, or otherwise, without the prior written permission of Wyatt Technology Corporation. ® DynaPro, DYNAMICS, Wyatt Technology, and its logo are registered trademarks of the Wyatt Technology Corporation. ™ Plate Reader and Protein Solutions are trademarks of Wyatt Technology Corporation. A variety of U.S. and foreign patents have been issued and/or are pending on various aspects of the apparatus and methodology implemented by this instrumentation. Table of Contents Chapter 1: Introduction Overview ............................................................................................................................6 The Instrument ............................................................................................................6 -
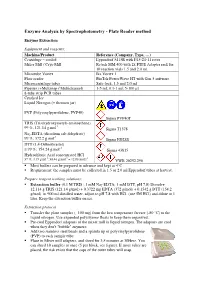
Enzyme Analysis by Spectrophotometry - Plate Reader Method
Enzyme Analysis by Spectrophotometry - Plate Reader method Enzyme Extraction Equipment and reagents: Machine/Product Reference (Company, Type, …) Centrifuge – cooled Eppendorf 5415R with F45-24-11 rotor Mixer Mill / Cryo Mill Retsch MM 400 with 2x PTFE Adapter rack for 10 reaction vials 1.5 and 2.0 ml. Microtube Vortex Ika Vortex 1 Plate reader BioTek PowerWave HT with Gen 5 software Microcentrifuge tubes Safe-lock, 1.5 and 2.0 ml Pipettes (+Multistep / Multichannel) 1-5 ml; 0.1-1 ml; 5-100 µl 8-tube strip PCR tubes Crushed Ice Liquid Nitrogen (+ thermos jar) PVP (Polyvinylpyrrolidone, PVP40) Sigma PVP40T TRIS (Tris(hydroxymetyl)-aminoethane) -1 99 %, 121.14 g.mol Sigma T1378 Na 2-EDTA (disodium salt dehydrate) -1 99 %; 372.2 g.mol Sigma ED2SS DTT (1,4-Dithiothreitol) -1 ≥ 99 %; 154.24 g.mol Sigma 43815 Hydrochloric Acid concentrated HCl -1 -1 -1 37 %; 1,19 g.ml ; 36.46 g.mol = 12.08 mol.l VWR 20252.290 Most buffers can be prepared in advance and kept at 4°C Requirement: the samples must be collected in 1.5 or 2.0 ml Eppendorf tubes at harvest. Prepare reagent working solutions: Extraction buffer (0.1 M TRIS ; 1 mM Na 2-EDTA; 1 mM DTT, pH 7.8) Dissolve 12.114 g TRIS (121.14 g/mol) + 0.3722 mg EDTA (372 g/mol) + 0.1542 g DTT (154.2 g/mol) in 900 ml distilled water, adjust to pH 7.8 with HCl (use 5M HCl) and dilute to 1 liter. Keep the extraction buffer on ice. -

Biotek's Most Comprehensive Cell Imaging Multi-Mode Reader
cell imagingimaging multi-mode reader reader BioTek’s most comprehensive cell imaging multi-mode reader. Comprehensive imaging solution cell imagingimaging multi-mode reader reader Cytation 7 is BioTek’s most comprehensive imaging plate reader with both inverted and upright microscopy enabling a wide range of applications in a compact and easy-to-use instrument. Cytation 7’s inverted microscopy module supports fluorescence, brightfield and color brightfield from 1.25x to 60x to analyze both large objects and intracellular details. Multi-mode plate reader with sophisticated imaging Cytation 7’s upright reflected light imaging module enables a broad range of applications such as ELISpot, colony counting, material inspection, and much more. Hit-picking: Multi-mode detection + imaging Cytation 7 builds on the legacy of the BioTek line of Synergy and Cytation readers with modular and upgradeable modes. Cytation 7 includes both upright and inverted microscopy optics which opens up a wide range of cellular and reflected light applications that cannot be performed on a standard plate reader. Information on cell morphology, localization of signal, cell count, object identification and quantification can be obtained with Cytation 7’s imaging modes. The monochromator plate reader optics allows running all of the standard plate reader assays. (1) Plate reader quickly identifies GFP positive wells. (2) Only GFP positive wells are imaged, saving both time and computer memory. Ready for any assay ELISpot Imaging Cytation 7’s upright imaging module can be used to automate assays such as ELISpot, in which cell With its combination of flexible plate reader and advanced microscopy mode, Cytation 7 is truly ready for secretions are rendered visible through the use of a colorimetric reaction. -

ELISA Technical Guide Table of Contents
ELISA Technical Guide Table of contents Introduction ................................................................................................................3 ELISA technology ......................................................................................................4 ELISA components ....................................................................................................6 ELISA equipment .......................................................................................................7 Equipment maintenance and calibration ..................................................................8 Reagent handling and preparation ...........................................................................9 Test component handling and preparation .............................................................10 Quality control .........................................................................................................11 Sample handling ......................................................................................................12 Pipetting methods ...................................................................................................14 ELISA plate timing ...................................................................................................16 ELISA plate washing ................................................................................................17 Plate reading and data management ......................................................................19 ELISA -

Modulus™ II Microplate Multimode Reader
Modulus™ II Microplate Multimode Reader Operating Manual Part Number 998-9375 Rev E Turner BioSystems, Inc. and its suppliers own the written and other materials in the Modulus II Microplate Reader operating manual. The written materials are copyrighted and protected by the copyright laws of the United States of America and, throughout the world, by the copyright laws of each jurisdiction according to pertinent treaties and conventions. No part of this manual may be reproduced manually, electronically, or in any other way without the written permission of Turner BioSystems, Inc. Modulus™ is a trademark of Turner BioSystems, Inc. All of the trademarks, service marks, and logos used in these materials are the property of Turner BioSystems, Inc. or their respective owners. All goods and services are sold subject to the terms and conditions of sale of the company within the Turner BioSystems, Inc. group that supplies them. A copy of these terms and conditions is available on request. NOTICE TO PURCHASER: The Modulus™ II Microplate Reader is for research purposes only. It is not intended or approved for diagnosis of disease in humans or animals. © Turner BioSystems, Inc. 2008 - All rights reserved. Made in the USA 2 Table of Contents 1 Instrument ............................................................................................................................... 7 1.1 Getting Started ................................................................................................................. 7 1.1.1 Description ........................................................................................................... -

PLATE READERS Increasing Throughput in Diagnostics
1/2 Infrared Temperature Sensors CASE STUDY PLATE READERS Increasing throughput in diagnostics Plate Reader Tecan Microplate Readers Tecan, founded in Switzerland in 1980, is a leading One of the products Tecan develops and global provider of laboratory instruments and manufactures are microplate readers. Their solutions. The company has manufacturing, cutting edge bench-top microplate readers and research and development sites in Europe washers lead the industry in both versatility and and North America and maintains a sales and performance. They offer single and multimode service network in 52 countries. They specialize microplate readers for all detection techniques. in development, production and distribution of The Spark 20M is their top class microplate automated workflow solutions for laboratories reader offering high performance for demanding in biopharmaceuticals, forensics and clinical applications in drug discovery and live sciences diagnostics. research. It offers total freedom of wavelengths selection and instant access to new wavelengths as assays requirements change. The modular architecture is ideally suited to researchers as well as service providers, allowing customers to configure the instrument to their budget and detection demands. Challenge Detection modes for microplate assays are absorbance, fluorescence intensity, luminescence, time-resolved fluorescence, and fluorescence polarization. All modalities are employed with the same objective: Measuring the exact concentration of the substance of interest. As some assays (e.g. AlphaScreen) are temperature sensitive, they need to be done within a narrow temperature range. Therefore, controlling the temperature of the microplate during the assay is essential, especially measuring the exact temperature of the liquid sample in every single well SEPARATELY. Normally, controlling the temperature of the microtiterplate is only done indirectly by controlling the ambient temperature in the plate reader and by controlling the temperature of the heating plate. -
PLATE READER INTRODUCTION Selecting the Right Plate Reader for Your Lab Can Be Challenging
CHOOSING THE RIGHT PLATE READER INTRODUCTION Selecting the right plate reader for your lab can be challenging. There are multiple factors to consider including: detection type, additional features, support, and price. With numerous options for each variable, it can be confusing to navigate the selection process. Focusing on striking a balance between functionality, optimization, and growth potential requires keen insight and continued vendor support throughout the life cycle of the plate reader. This detailed guide will help you choose the right plate reader for your needs by examining the marketplace and pointing out considerations to keep in mind when making your decision. UNDERSTANDING THE PROCESS A few key questions can help customers navigate toward the best fit for a reader meeting the individual specification requirements for your lab: What detection technology should you look for when selecting a plate reader for your lab? Is this technology ideal for multiple therapeutic areas? How can you get the best performance optimization out of your reader? What support options does your vendor offer in conjunction with the purchase? WHAT DETECTION TECHNOLOGY SHOULD YOU LOOK FOR WHEN SELECTING A PLATE READER FOR YOUR LAB? Whether a lab performs development, high-throughput screening, or a more specialized area of focus, a reader should offer, at minimum, the requirements needed for each individual laboratory. For example, a growing lab focused on performing reporter gene assays requires a plate reader that delivers on the importance of sensitivity while offering the potential for future upgrades or additional features as the lab matures over time. Vendors with reader option focused on providing sensitivity, speed, and multiplex capabilities can meet the sensitivity needs of reporter gene assay performance while offering additional reading technologies for customization and accommodating the fluctuation of industry trends in support of long-term growth. -

An Open-Source Plate Reader Karol Szymula University of Pennsylvania
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by ScholarlyCommons@Penn University of Pennsylvania ScholarlyCommons Departmental Papers (BE) Department of Bioengineering 2018 An Open-Source Plate Reader Karol Szymula University of Pennsylvania Michael S. Magaraci University of Pennsylvania, [email protected] Michael Patterson University of Pennsylvania Andrew Clark University of Pennsylvania Sevile G. Mannickarottu University of Pennsylvania See next page for additional authors Follow this and additional works at: https://repository.upenn.edu/be_papers Part of the Biomedical Engineering and Bioengineering Commons Recommended Citation Szymula, K., Magaraci, M. S., Patterson, M., Clark, A., Mannickarottu, S. G., & Chow, B. Y. (2018). An Open-Source Plate Reader. Retrieved from https://repository.upenn.edu/be_papers/221 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/be_papers/221 For more information, please contact [email protected]. An Open-Source Plate Reader Abstract Microplate readers are foundational instruments in experimental biology and bioengineering that enable multiplexed spectrophotometric measurements. To enhance their accessibility, we here report the design, construction, validation, and benchmarking of an open-source microplate reader. The system features full- spectrum absorbance and fluorescence emission detection, in situ optogenetic stimulation, and stand-alone touch screen programming of automated assay protocols. The ott al system costs Keywords open source, low cost hardware, plate reader, synthetic biology, high throughput Disciplines Biomedical Engineering and Bioengineering | Engineering Author(s) Karol Szymula, Michael S. Magaraci, Michael Patterson, Andrew Clark, Sevile G. Mannickarottu, and Brian Y. Chow This working paper is available at ScholarlyCommons: https://repository.upenn.edu/be_papers/221 An Open-Source Plate Reader Karol Szymula,† Michael S. -

Faqs for Laboratory Professionals Quantiferon®-TB Gold Plus
FAQs for Laboratory Professionals QuantiFERON®-TB Gold Plus Sample to Insight Contents Questions and answers 3 Test principle 3 Blood collection 4 The blood hasn’t reached the black mark on the side of the QFT-Plus blood collection tube. Is this important? 4 How important is the tube mixing process? 4 Can the blood collection tubes be transported lying down? 4 Blood incubation / plasma harvesting 4 What if 37ºC incubation starts more than 16 hours after the time of blood collection for specimens collected directly into QFT-Plus blood collection tubes? 4 Can I incubate the blood collection tubes lying down? 5 Do I have to centrifuge the tubes before I can harvest the plasma? 5 Do I have to centrifuge the tubes immediately after removal from the incubator? 5 The gel plug hasn’t moved during centrifugation. What should I do? 5 The plasma doesn’t appear the color it normally does. Is this OKAY? 5 What volume of plasma do I need to harvest from above the sedimented red blood cells or gel plug? Is this important? 6 I want to maximize the cost-effectiveness of the QuantiFERON-TB Gold Plus assay by batching my samples. What is the stability associated with the harvested plasma? 6 What should I do if clots form in my plasma samples during frozen storage? 6 Do I need to use microtubes when storing harvested plasma? Can I use more cost-effective microtiter plates in this instance? 6 Interferon-gamma (IFN-γ) ELISA 6 What is the stability associated with these selected kit components— 6 Can I use the QuantiFERON ELISA plate immediately after removal from the refrigerator? 7 Do I require an automated Plate Washer? 7 How important is washing during the QuantiFERON ELISA? 7 2 QuantiFERON-TB Gold Plus FAQs for Laboratory Professionals 03/2018 Data analysis 7 I have very high Nil control values? What may be the problem? 7 A patient’s TB Antigen value is very high (possibly above the detectable limit of the plate reader). -

Multiwell Solutions for Discovery Research and Sample Prep
Multiply your success. Multiwell solutions for discovery research and sample prep. 01 Sample Preparation 3 Table of Contents Microporous Sample Prep 3 This guide for discovery research, molecular biology, Resin-based Separation 6 and sample preparation includes products for high- throughput screening and cell culture. These products Genomics Sample Prep 7 provide proven solutions for a range of applications Sequencing Reaction Cleanup 9 and are backed by extensive technical support. Protein Concentration and Desalting 10 For more information 02 Biochemical Assays 11 For more information on products and technical Enzyme Assays 11 support, visit www.millipore.com/cellculture Receptor Binding Assays 14 03 Cell-based Assays 16 Membrane-based Cell Assays 16 Tissue Culture Treated Plates 21 Migration, Invasion and Chemotaxis 22 Toxicity Using Whole Organism Models 26 Elispot Assays 27 04 ADME/Compound Profiling 29 Aqueous Solubility 29 Non Cell-based Absorption 33 05 Accessories 34 Vacuum Filtration 36 Millicell® ERS-2 Voltohmmeter 37 Radiometric Assays 38 Centrifugation and Chromatography 38 Collection Plates 38 06 Appendix 39 ® MultiScreen HTS Filter Plate Selection Guide 39 Sample Preparation 3 Microporous Sample Prep 3 01 Sample Preparation Resin-based Separation 6 Genomics Sample Prep 7 Multiwell plates enable simple, rapid, automation-compatible sample preparation within the life science, Sequencing Reaction Cleanup 9 environmental analysis, clinical, forensic and industrial quality control markets, meeting the demands of Protein Concentration and Desalting 10 lower detection limits and higher throughput. MultiScreen® filter plates meet specific performance criteria for sample preparation methodology requiring Biochemical Assays 11 low nonspecific binding of protein and drug analytes, solvent compatibility and sample throughput.